- UroToday Home

- Library Resources
- Localized Prostate Cancer
Library Resources
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Huggins, Charles, and Clarence V. Hodges. "Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate." The Journal of urology 167, no. 2 Part 2 (2002): 948-951.
2. Coutinho, Isabel, Tanya K. Day, Wayne D. Tilley, and Luke A. Selth. "Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence." Endocrine-related cancer 23, no. 12 (2016): T179-T197.
3. Visakorpi, Tapio, Eija Hyytinen, Pasi Koivisto, Minna Tanner, Riitta Keinänen, Christian Palmberg, Aarno Palotie, Teuvo Tammela, Jorma Isola, and Olli-P. Kallioniemi. "In vivo amplification of the androgen receptor gene and progression of human prostate cancer." Nature genetics 9, no. 4 (1995): 401-406.
4. Chen, Charlie D., Derek S. Welsbie, Chris Tran, Sung Hee Baek, Randy Chen, Robert Vessella, Michael G. Rosenfeld, and Charles L. Sawyers. "Molecular determinants of resistance to antiandrogen therapy." Nature medicine 10, no. 1 (2004): 33-39.
5. Wyatt, Alexander W., and Martin E. Gleave. "Targeting the adaptive molecular landscape of castration‐resistant prostate cancer." EMBO molecular medicine 7, no. 7 (2015): 878-894.
6. Robinson, Dan, Eliezer M. Van Allen, Yi-Mi Wu, Nikolaus Schultz, Robert J. Lonigro, Juan-Miguel Mosquera, Bruce Montgomery et al. "Integrative clinical genomics of advanced prostate cancer." Cell 161, no. 5 (2015): 1215-1228.
7. Grasso, Catherine S., Yi-Mi Wu, Dan R. Robinson, Xuhong Cao, Saravana M. Dhanasekaran, Amjad P. Khan, Michael J. Quist et al. "The mutational landscape of lethal castration-resistant prostate cancer." Nature 487, no. 7406 (2012): 239-243.
8. Cai, Changmeng, Housheng Hansen He, Sen Chen, Ilsa Coleman, Hongyun Wang, Zi Fang, Shaoyong Chen et al. "Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1." Cancer cell 20, no. 4 (2011): 457-471.
9. Antonarakis, Emmanuel S., Changxue Lu, Hao Wang, Brandon Luber, Mary Nakazawa, Jeffrey C. Roeser, Yan Chen et al. "AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer." New England Journal of Medicine 371, no. 11 (2014): 1028-1038.
10. Chmelar, Renée, Grant Buchanan, Eleanor F. Need, Wayne Tilley, and Norman M. Greenberg. "Androgen receptor coregulators and their involvement in the development and progression of prostate cancer." International Journal of cancer 120, no. 4 (2007): 719-733.
11. Zoubeidi, Amina, Anousheh Zardan, Eliana Beraldi, Ladan Fazli, Richard Sowery, Paul Rennie, Colleen Nelson, and Martin Gleave. "Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity." Cancer research 67, no. 21 (2007): 10455-10465.
12. Gregory, Christopher W., Bin He, Raymond T. Johnson, O. Harris Ford, James L. Mohler, Frank S. French, and Elizabeth M. Wilson. "A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy." Cancer research 61, no. 11 (2001): 4315-4319.
13. Qin, Jun, Hui-Ju Lee, San-Pin Wu, Shih-Chieh Lin, Rainer B. Lanz, Chad J. Creighton, Francesco J. DeMayo, Sophia Y. Tsai, and Ming-Jer Tsai. "Androgen deprivation–induced NCoA2 promotes metastatic and castration-resistant prostate cancer." The Journal of clinical investigation 124, no. 11 (2014): 5013-5026.
Bone-Targeted Therapy in Prostate Cancer
Zoledronic acid
To maintain bone integrity during bone remodeling, homeostasis of osteoblasts increasing bone mass and osteoclasts resorbing bone is required. Bisphosphonates are rapidly absorbed on the bone surface and inhibit osteoclast activity by affecting cytoskeletal dynamics. The Phase III Zoledronic acid 039 trial showed that among men with metastatic castration-resistant prostate cancer (mCRPC), a greater proportion of patients who received placebo had skeletal-related events than those who received zoledronic acid at 4 mg (44.2% versus 33.2%, p =0.021) or those who received zoledronic acid at 8 mg (38.5%, p = 0.222- Written by: Zachary Klaassen, MD, MSc
- References:
1. Norgaard M, Jensen AO, Jacobsen JB, Cetin K, Fryzek JP, Sorensen HT. Skeletal related events, bone metastasis and survival of prostate cancer: a population based cohort study in Denmark (1999 to 2007). J Urol. 2010;184(1):162-167.
2. Gartrell BA, Coleman R, Efstathiou E, et al. Metastatic Prostate Cancer and the Bone: Significance and Therapeutic Options. Eur Urol. 2015;68(5):850-858.
3. Klaassen Z, Howard LE, de Hoedt A, et al. Factors predicting skeletal-related events in patients with bone metastatic castration-resistant prostate cancer. Cancer. 2017;123(9):1528-1535.
4. Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94(19):1458-1468.
5. Smith MR, Halabi S, Ryan CJ, et al. Randomized controlled trial of early zoledronic acid in men with castration-sensitive prostate cancer and bone metastases: results of CALGB 90202 (alliance). J Clin Oncol. 2014;32(11):1143-1150.
6. Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377(9768):813-822.
7. Smith MR, Saad F, Oudard S, et al. Denosumab and bone metastasis-free survival in men with nonmetastatic castration-resistant prostate cancer: exploratory analyses by baseline prostate-specific antigen doubling time. J Clin Oncol. 2013;31(30):3800-3806.
8. Heidenreich A, Bastian PJ, Bellmunt J, et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2014;65(2):467-479.
9. Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-223.
10. Smith M, Parker C, Saad F, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(3):408-419.
11. Vignani F, Bertaglia V, Buttigliero C, Tucci M, Scagliotti GV, Di Maio M. Skeletal metastases and impact of anticancer and bone-targeted agents in patients with castration-resistant prostate cancer. Cancer Treat Rev. 2016;44:61-73.
12. Basch E, Autio KA, Smith MR, et al. Effects of cabozantinib on pain and narcotic use in patients with castration-resistant prostate cancer: results from a phase 2 nonrandomized expansion cohort. Eur Urol. 2015;67(2):310-318.
13. Araujo JC, Mathew P, Armstrong AJ, et al. Dasatinib combined with docetaxel for castration-resistant prostate cancer: results from a phase 1-2 study. Cancer. 2012;118(1):63-71.
14. Araujo JC, Trudel GC, Saad F, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol. 2013;14(13):1307-1316.
15. Nelson JB. Endothelin receptor antagonists. World J Urol. 2005;23(1):19-27.
16. Nelson JB, Love W, Chin JL, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer. 2008;113(9):2478-2487.
First Line Therapy for Metastatic Clear Cell Renal Cell Carcinoma
Second perhaps only to advanced prostate cancer, the metastatic clear cell renal cell carcinoma disease space has undergone rapid and transformational change over the past fifteen years. This rapidly shifting treatment landscape was highlighted recently at the American Society of Clinical Oncology 2019 Annual Meeting:
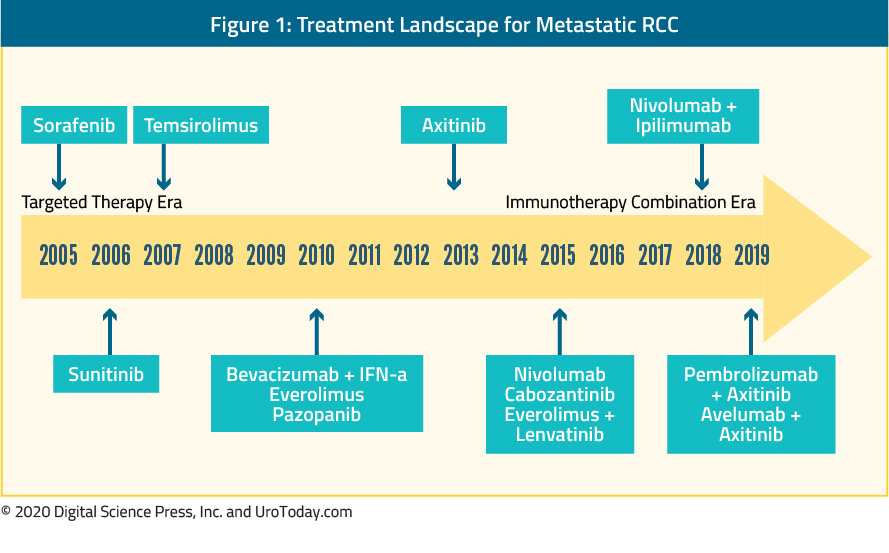
Early years: cytokine therapy
It has been recognized for many decades that renal cell carcinoma is an immunologically active tumor. As a result, modulators of the immune system were among the first therapeutic targets for advanced ccRCC. Prior to 2005, treatment for metastatic RCC (mRCC) was limited to cytokine therapies (interferon-alfa and interleukin-2).
Interferon-α was one of the first cytokines assessed for the treatment of metastatic ccRCC. Based on early data suggesting a response rate between 10 to 15%5 and comparative data demonstrating a survival benefit compared to other available systemic therapies available at the time,6 interferon-alfa retained utilization despite significant toxicity. Further, it was among patients with metastatic RCC receiving interferon-alfa that the Motzer prognostic criteria were derived.6 In their seminal paper in the Journal of Clinical Oncology, Motzer and colleagues demonstrated that low Karnofsky performance status, high lactate dehydrogenase, low serum hemoglobin, high corrected serum calcium, and short time from initial RCC diagnosis to start of interferon-alfa therapy (<1 year) could be used to risk-stratify patients with renal cell carcinoma. However, even among patients treated at a center of excellence, median overall survival was only 30 months in favorable-risk patients, 14 months in intermediate-risk patients and five months in poor-risk patients.6
Other immunologic therapies were explored including interleukin-2. While response rates were similar to interferon-based therapies (~15 to 20%),7 interleukin-2 was distinct in that durable complete responses were observed in approximately 7 to 9% of patients.8 This observation led to the U.S. Food and Drug Administration (FDA) approval of high-dose IL-2 in 1992. However, IL-2 is associated with significant toxicity which has limited its widespread use.
Combinations of interferon and interleukin therapies were explored subsequently those these data demonstrated no improvement in overall survival,9 with significantly increased toxicity compared to monotherapy with either agent.
A new standard: molecularly targeted agents
Based on work into the molecular biology underlying ccRCC, researchers were led to “rational targeted therapeutics” including targeting of the vascular endothelial growth factor (VEGF) pathway and mammalian target of rapamycin (mTOR). Mammalian target of rapamycin (mTOR) plays a key role in regulating HIF-α, thus modulating the pathway between abnormalities in VHF and proliferation.
Bevacizumab, a humanized monoclonal antibody against VEGF-A, was the first inhibitor of the VEGF pathway used in clinical trials. As is standard in an oncology pathway, it was first tested in patients who had progressed on the current standard of care (cytokine therapy) and subsequently tested in untreated patients. In head-to-head trials against interferon-alfa, the addition of bevacizumab to interferon resulted in significant improvements in response rate and progression-free survival.10,11 Today, bevacizumab is uncommonly used as monotherapy in untreated patients but is considered as second-line therapy in patients who have failed prior therapy with tyrosine kinase inhibitors.
Tyrosine-kinase inhibitors also target the VEGF pathway, through inhibition of a combination of VEGFR-2, PDGFR-β, raf-1 c-Kit, and Flt3 (sunitinib and sorafenib). In 2006, sorafenib was shown to have biologic activity in ccRCC. Subsequent studies demonstrated improvements in progression-free survival compared with placebo in patients who have previously failed cytokine therapy and improvements in tumor regression compared to interferon in previously untreated patients. As highlighted in the Figure above, sorafenib was one the first molecularly targeted agents clinically available. However, despite FDA approval, sorafenib was quickly supplanted by sunitinib as a first-line VEGF inhibitor.
In keeping with the aforementioned oncology pipeline, sunitinib was first evaluated among patients who had previously received cytokine treatment. Subsequently, it was compared to interferon-α in a pivotal Phase III randomized trial.12 Among 750 patients with previously untreated, metastatic RCC randomized, median progression-free survival was significantly longer among those who received sunitinib (11 months) than those who received interferon-alfa (5 months; hazard ratio 0.42, 95% confidence interval 0.32 to 0.54). Similar benefits were seen in the overall response rate with subsequent follow-up demonstrating a strong trend towards improved overall survival. In the pivotal trial, patients who received sunitinib had a significantly better quality of life than those who received interferon-alfa12, despite class-based toxicity profile including gastrointestinal events, dermatologic complications including hand-foot desquamation, hypertension, and general malaise. On account of these data, sunitinib is widely used as a first-line treatment of RCC.
Since the approval of sunitinib and sorafenib, there has been the development and subsequent approval of a number of other tyrosine kinase inhibitors. For the most part, the goal of these agents has been to reduce the toxicity of VEGF inhibitors while retaining oncologic efficacy. Comparative data of pazopanib and sunitinib have demonstrated non-inferior oncologic outcomes with decreased toxicity among patients receiving pazopanib.13 Axitinib was evaluated first as second-line therapy14 and then in the first-line setting compared to sorafenib.15 Among 192 patients with previously untreated ccRCC randomized to axitinib and 96 patients randomized to sorafenib, median progression-free survival was not significantly different (10.1 months and 6.5 months, respectively; hazard ratio 0.77, 95% confidence interval 0.56 to 1.05)15. Finally, tivozanib has been compared to sorafenib among patients who had not previously received VEGF or mTOR-targeting therapies. While this study demonstrated tivozanib’s activity, it was not FDA approved and is therefore not used.
Most recently, a multikinase inhibitor, cabozantinib has been approved for the first-line treatment of mRCC. In the Phase II CABOSUN trial, cabozantinib was compared to sunitinib in the first-line treatment of patients with intermediate or poor-risk mRCC.16 Assessing the primary outcome of progression-free survival, the 79 patients randomized to cabozantinib had significantly longer progression-free survival (8.2 months) compared to the 78 randomized to sunitinib (5.6 months; hazard ratio 0.66, 95% confidence interval 0.46 to 0.95). A recent update on this trial utilizing independent progression-free survival (PFS) review demonstrated comparable results (hazard ratio 0.48, 95% confidence interval 0.31 to 0.74)17. Even with an increased follow-up (median 34.5 months), no significant difference in overall survival was demonstrated (26.6 months in patients receiving cabozantinib and 21.2 months in those receiving sunitinib; hazard ratio 0.80, 95% confidence interval 0.53 to 1.21). While this appears to demonstrate a significant benefit to cabozantinib, median survival in the sunitinib arm was lower than may be expected18 which would serve to exaggerate the apparent benefit of cabozantinib.
In parallel to the development, clinical appraisal and utilization of VEGF inhibitors have come the development of mTOR inhibitors. Temsirolimus was the first mTOR inhibitor to reach clinical utility in patients with metastatic RCC. In the Global ARCC Trial, temsirolimus, interferon, and the combination were compared among 626 patients with pre-defined poor-risk metastatic RCC who had not previously received systemic therapy.19 Notable compared to many trials in this disease space that have utilized progression-free survival as the primary outcome, overall survival was the primary outcome, with the study powered based on comparisons of the temsirolimus group and the combination group to the interferon-alfa group. Patients who received temsirolimus had significantly improved overall survival compared to those receiving interferon-alfa (hazard ratio 0.73, 95% confidence interval 0.58 to 0.92). Notably, the combination arm did not offer a benefit compared to interferon alone. Unlike temsirolimus which must be administered intravenously, everolimus is an oral agent.
What’s old is new: immunotherapy for RCC
The immunologic basis for the treatment of advanced RCC has been well established, including the aforementioned cytokine therapies. Thus, it should not be surprising that the use of checkpoint inhibitors has demonstrated benefit in patients with metastatic RCC.
First presented at ESMO in the fall of 2017 and subsequently published in the spring of 2018, CheckMate 214 demonstrated an overall survival (OS) benefit for first-line nivolumab plus ipilimumab vs sunitinib.20 This trial randomized 1096 patients to the combination immunotherapy approach of nivolumab plus ipilimumab (550 patients) or sunitinib (546 patients). The majority of patients had intermediate or poor-risk disease (n=847). Overall survival was significantly improved in the overall patient population; however, stratified analyses provide more nuanced results. Among the subgroup of patients with intermediate or poor-risk RCC, treatment with nivolumab plus ipilimumab resulted in significantly improved overall response rate, comparable progression-free survival, and significantly improved overall survival. In contrast, among patients with favorable-risk disease, progression-free survival and overall response rate were higher among patients who received sunitinib. Recently, Escudier and colleagues have assessed the efficacy of nivolumab and ipilimumab according to the number of IMDC risk factors.21 In keeping with the previously reported differences in the comparative benefit of nivo/ipi versus sunitinib on the basis of risk category (intermediate/poor versus favorable), the authors demonstrated stable overall response rate (ORR) across increasing numbers of IMDC risk factors (from zero to six) for those who received nivolumab and ipilimumab, while the ORR in patients treated with sunitinib decreased with an increasing number of IMDC risk factors.
The next frontier: combinations of targeted therapy and immunotherapy
Shortly after the data from CheckMate214 emerged, the results of IMmotion151 were presented at GU ASCO in the spring of 2018 and subsequently published. This Phase III trial compared first-line atezolizumab + bevacizumab versus sunitinib among 915 patients with previously untreated metastatic RCC.22 This regime was active with a significant benefit in progression-free survival (11.2 months versus 7.7 months; hazard ratio 0.74, 95% confidence interval 0.57 to 0.96) among the whole cohort of patients and had lower rates of significant (grade 3-4) adverse events (40% vs 54%).
Since the publication of CheckMate214 and IMmotion151, two trials have reported on combinations of checkpoint inhibitors and tyrosine kinase inhibitors: KEYNOTE-426 and JAVELIN Renal 101.
In KEYNOTE-426, 861 patients with metastatic clear cell RCC who had not previously received systemic therapy were randomized to pembrolizumab plus axitinib or sunitinib and followed for the co-primary endpoints of overall survival and progression-free survival.23 Similar to CheckMate214, the majority of patients had intermediate or poor-risk disease. While median survival was not reached, patients who received pembrolizumab and axitinib had improved overall survival (hazard ratio 0.53, 95% confidence interval 0.38 to 0.74) and progression-free survival (hazard ratio 0.69, 95% confidence interval 0.57 to 0.84), as well as overall response rate. These results were consistent across subgroups of demographic characteristics, IMDC risk categories, and programmed death-ligand 1 (PD-L1) expression level. Grade 3 to 5 adverse events were somewhat more common among patients getting pembrolizumab and axitinib, though rates of discontinuation were lower.
Similarly, JAVELIN Renal 101 randomized 886 patients to avelumab and axitinib or sunitinib.24 Again, the preponderance of patients had IMDC intermediate or poor-risk disease. This analysis of the primary endpoints was progression-free survival and overall survival in patients with PD-L1 positive tumors. Notably, 560 of the 886 patients had PD-L1 positive tumors. Among the PD-L1 positive subgroup, progression-free survival (hazard ratio 0.61, 95% confidence interval 0.47 to 0.79) was improved in patients receiving avelumab and axitinib compared to sunitinib while overall survival did not significantly differ (hazard ratio 0.82, 95% confidence interval 0.53 to 1.28). In the overall study population, progression-free survival was similarly improved, as compared to the PD-L1 positive population (hazard ratio 0.69, 95% confidence interval 0.56 to 0.84).
Dead-ends
Numerous chemotherapeutic agents have been explored in ccRCC. These include 5-FU, gemcitabine, vinblastine, bleomycin, and platinum. Meta-analyses of these data demonstrate poor response25 and thus cytotoxic chemotherapy is not indicated in the treatment of advanced RCC. Similarly, hormonal therapies including medroxyprogesterone have been explored but have no role in the modern management of advanced RCC.
Integration of treatment options for patients with metastatic ccRCC
Due to the rapid proliferation of treatment options in first-line treatment of metastatic clear cell renal cell carcinoma, there is a paucity of direct comparative data. The majority of new agents have been compared to sunitinib which was the standard of care at the time that trials were designed. Due to the lack of comparative data, it may be difficult to ascertain which treatment to offer patients who present in clinic. There are a number of ways to approach this issue. First, one may take a quantitative approach, utilizing the available comparative data in a network meta-analysis; second, one may rely upon eminence, as in expert-informed guidelines; finally, one may rely on individual clinical experience.
Assessing this quantitatively, we have recently performed a network meta-analysis of first-line agents in metastatic RCC.26 Assessing agents which are commonly utilized in 2019, we examined 12 relevant trials. Depending on the outcome of interest (progression-free survival, overall survival, or adverse events), the preferred treatment varied. However, pembrolizumab and axitinib appeared to have a high likelihood of being preferred for oncologic outcomes.
Second, considering a panel of expert opinion, the European Association of Urology recently updated its guidelines on the treatment of renal cell carcinoma. Their recommendations are highlighted in the following figure, taken from the EAU guideline:
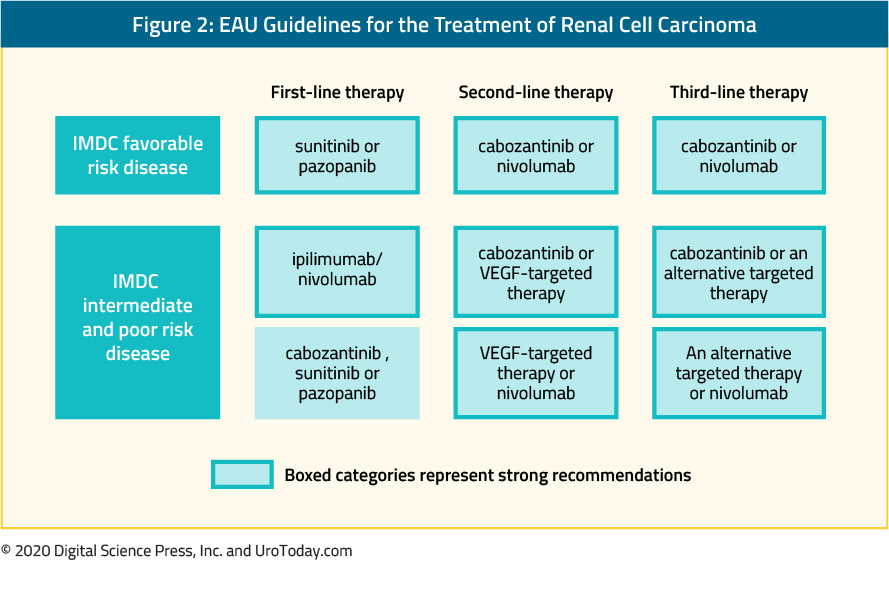
Notably, the most recent version of these guidelines alludes to the recently published data but have not yet integrated the role of atezolizumab plus bevacizumab, pembrolizumab plus axitinib, or avelumab plus axitinib in guideline recommendations.
Finally, we may rely on the guidance of individual clinical experience.
What about surgery?
The role of cytoreductive nephrectomy in the management of metastatic renal cell carcinoma has dramatically changed with the publication of the CARMENA and SURTIME studies. The available evidence suggests that systemic therapy should be prioritized ahead of cytoreductive nephrectomy. However, there remains a role of cytoreductive nephrectomy in select patients.
Published Date: March 17th, 2020
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians. 2018;68(1):7-30.
2. Welch HG, Skinner JS, Schroeck FR, Zhou W, Black WC. Regional Variation of Computed Tomographic Imaging in the United States and the Risk of Nephrectomy. JAMA internal medicine. 2018;178(2):221-227.
3. Motzer RJ, Mazumdar M, Bacik J, Berg W, Amsterdam A, Ferrara J. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. Journal of Clinical Oncology. 1999;17:2530-2540.
4. Negrier S, Escudier B, Gomez F, et al. Prognostic factors of survival and rapid progression in 782 patients with metastatic renal carcinomas treated by cytokines: a report from the Groupe Francais d'Immunotherapie. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2002;13(9):1460-1468.
5. Motzer RJ, Bacik J, Murphy BA, Russo P, Mazumdar M. Interferon-alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2002;20(1):289-296.
6. Coppin C, Porzsolt F, Awa A, Kumpf J, Coldman A, Wilt T. Immunotherapy for advanced renal cell cancer. Cochrane Database Syst Rev. 2005(1):CD001425.
7. Dutcher JP, Atkins M, Fisher R, et al. Interleukin-2-based therapy for metastatic renal cell cancer: the Cytokine Working Group experience, 1989-1997. Cancer J Sci Am. 1997;3 Suppl 1:S73-78.
8. Rosenberg SA, Yang JC, White DE, Steinberg SM. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: identification of the antigens mediating response. Ann Surg. 1998;228(3):307-319.
9. Negrier S, Escudier B, Lasset C, et al. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. Groupe Francais d'Immunotherapie. The New England journal of medicine. 1998;338(18):1272-1278.
10. Rini BI, Halabi S, Rosenberg JE, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(33):5422-5428.
11. Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370(9605):2103-2111.
12. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. The New England journal of medicine. 2007;356(2):115-124.
13. Motzer RJ, Hutson TE, Cella D, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. The New England journal of medicine. 2013;369(8):722-731.
14. Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378(9807):1931-1939.
15. Hutson TE, Lesovoy V, Al-Shukri S, et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomised open-label phase 3 trial. The lancet oncology. 2013;14(13):1287-1294.
16. Choueiri TK, Halabi S, Sanford BL, et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017;35(6):591-597.
17. Choueiri TK, Hessel C, Halabi S, et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. European journal of cancer. 2018;94:115-125.
18. Buti S, Bersanelli M. Is Cabozantinib Really Better Than Sunitinib As First-Line Treatment of Metastatic Renal Cell Carcinoma? Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017;35(16):1858-1859.
19. Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. The New England journal of medicine. 2007;356(22):2271-2281.
20. Escudier B, Tannir NM, McDermott D, et al. LBA5 - CheckMate 214: Efficacy and safety of nivolumab 1 ipilimumab (N1I) v sunitinib (S) for treatment-naive advanced or metastatic renal cell carcinoma (mRCC), including IMDC risk and PD-L1 expression subgroups. Annals of Oncology. 2017;28(Supplement 5):621-622.
21. Escudier B, Motzer RJ, Tannir NM, et al. Efficacy of Nivolumab plus Ipilimumab According to Number of IMDC Risk Factors in CheckMate 214. European urology. 2019.
22. Motzer R, Powles T, Atkins M, et al. IMmotion151: A Randomized Phase III Study of Atezolizumab Plus Bevacizumab vs Sunitinib in Untreated Metastatic Renal Cell Carcinoma. Journal of Clinical Oncology. 2018;36(Suppl 6S).
23. Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. The New England journal of medicine. 2019;380(12):1116-1127.
24. Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. The New England journal of medicine. 2019;380(12):1103-1115.
25. Yagoda A, Abi-Rached B, Petrylak D. Chemotherapy for advanced renal-cell carcinoma: 1983-1993. Semin Oncol. 1995;22(1):42-60.
26. Hahn AW, Klaassen Z, Agarwal N, et al. First-line Treatment of Metastatic Renal Cell Carcinoma: A Systematic Review and Network Meta-analysis. Eur Urol Oncol. 2019;2(6):708-715.
Adjuvant Systemic Therapy for High Risk Kidney Cancer
Adjuvant targeted therapy
Tyrosine kinase inhibitors (TKIs) quickly became standard of care for patients with metastatic renal cell carcinoma following their introduction in the early 2000s. They have subsequently been investigated as adjuvant therapy in 4 published randomized trials to our knowledge. In addition, the SORCE trial was presented at ESMO 2019 at the end of September 2019.
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Patel HD, Gupta M, Joice GA, et al. Clinical Stage Migration and Survival for Renal Cell Carcinoma in the United States. Eur Urol Oncol 2019; 2(4):343-348
2. Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet 2016; 387(10032):2008-16.
3. Motzer RJ, Haas NB, Donskov F, et al. Randomized Phase III Trial of Adjuvant Pazopanib Versus Placebo After Nephrectomy in Patients With Localized or Locally Advanced Renal Cell Carcinoma. J Clin Oncol 2017; 35(35):3916-3923.
4. Ravaud A, Motzer RJ, Pandha HS, et al. Adjuvant Sunitinib in High-Risk Renal-Cell Carcinoma after Nephrectomy. N Engl J Med 2016; 375(23):2246-2254.
5. Gross-Goupil M, Kwon TG, Eto M, et al. Axitinib versus placebo as an adjuvant treatment of renal cell carcinoma: results from the phase III, randomized ATLAS trial. Ann Oncol 2018; 29(12):2371-2378.
6. Haas NB, Manola J, Dutcher JP, et al. Adjuvant Treatment for High-Risk Clear Cell Renal Cancer: Updated Results of a High-Risk Subset of the ASSURE Randomized Trial. JAMA Oncol 2017; 3(9):1249-1252
7. Sun M, Marconi L, Eisen T, et al. Adjuvant Vascular Endothelial Growth Factor-targeted Therapy in Renal Cell Carcinoma: A Systematic Review and Pooled Analysis. Eur Urol 2018; 74(5):611-620.
8. Spek A, Szabados B, Casuscelli J, et al. Adjuvant therapy in renal cell carcinoma: the perspective of urologists. Int J Clin Oncol 2019; 24(6):694-697.
9. Martinez Chanza N, Tripathi A, Harshman LC. Adjuvant Therapy Options in Renal Cell Carcinoma: Where Do We Stand? Curr Treat Options Oncol 2019; 20(5):44.
10. Gleeson JP, Motzer RJ, Lee CH. The current role for adjuvant and neoadjuvant therapy in renal cell cancer. Curr Opin Urol 2019.
11. Ljungberg B, Albiges L, Abu-Ghanem Y, et al. European Association of Urology Guidelines on Renal Cell Carcinoma: The 2019 Update. Eur Urol 2019; 75(5):799-810.
12. Wood C, Srivastava P, Bukowski R, et al. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet 2008; 372(9633):145-54.
13. Aitchison M, Bray CA, Van Poppel H, et al. Final results from an EORTC (GU Group)/NCRI randomized phase III trial of adjuvant interleukin-2, interferon alpha, and 5-fluorouracil in patients with a high risk of relapse after nephrectomy for renal cell carcinoma (RCC). Journal of Clinical Oncology 2011; 29(15 (SUPPL)):4505.
14. Tsimafeyeu ID, L., Kharkevich G, Petenko N, et al. Granulocyte-Macrophage Colony-Stimulating Factor, Interferon Alpha and Interleukin-2 as Adjuvant Treatment for High-Risk Renal Cell Carcinoma. J Cancer Sci Ther 2010; 2:157-159.
15. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018; 378(14):1277-1290.
16. Motzer RJ, Penkov K, Haanen J, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019; 380(12):1103-1115.
17. Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019; 380(12):1116-1127.
Prevention of Skeletal-Related Events in Advanced Prostate Cancer
Patients with advanced prostate cancer are at significant risk of skeletal-related events (SREs) due to a complex interplay between bone health and prostate cancer due to cancer biology and the predilection of prostate cancer to spread to bone, the toxicity of prostate cancer treatments, and shared epidemiology of the two conditions.
Skeletal-related events are used to denote events related to osseous metastases, including pathologic bone fractures, spinal cord compression, orthopedic surgical intervention, and palliative radiation directed at the bone.1 This definition has been widely used in the design of randomized controlled trials and for drug approval. In some circumstances, authors include a change in systemic anti-neoplastic therapy as a result of bony pain in the definition of skeletal-related events. The key to this definition is that skeletal-related events may be clinically manifested due to symptoms or only radiographically detected. Thus, skeletal-related events may or may not be symptomatic. In contrast, symptomatic skeletal-related events (SSEs) is a relatively newer outcome representing a subset of skeletal-related events which symptomatically affect the patient experience. This outcome was first used in the Alpharadin in Symptomatic Prostate Cancer (ALSYMPCA) trial,2 wherein symptomatic skeletal-related events were defined as bone-directed radiotherapy to relieve bony pain, new symptomatic pathologic fractures, spinal cord compression, or tumor-related orthopedic surgery. While there is a significant overlap between these conditions, there are important differences that relate to both study design and patient care: namely, detection of SREs requires routine radiographic evaluation to detect asymptomatic skeletal-related events while detection of SSEs can be driven by patient evaluation.
Skeletal related events contribute significantly to the disease-related morbidity and mortality of prostate cancer, in addition to being very costly. A range of lifestyle, nutritional, and pharmaceutical interventions can be undertaken to decrease the risk of skeletal-related events in patients with advanced prostate cancer.
Lifestyle and nutrition-based interventions
These recommendations are not unique to patients with prostate cancer, but rather apply to all men with osteoporosis or decreased bone mineral density to reduce the risk of fractures.
The Endocrine Society Clinical Practice Guideline on Osteoporosis in Men recommends the following lifestyle interventions:3
- Calcium intake: it is recommended that men with or at risk for osteoporosis consume 1000 to 1200 mg of calcium daily. While dietary sources are preferable, calcium supplementation should be used if dietary calcium intake is inadequate.
- Vitamin D intake: men with low vitamin D levels (< 30 ng/mL or < 75 nmol/L) should receive vitamin D supplementation to raise serum vitamin D levels to at least these levels.
- Exercise: men at risk of osteoporosis are recommended to participate in weight-bearing activities at least three or four times per week, for 30 to 40 minutes per session.
- Alcohol intake: it is recommended that men at risk of osteoporosis reduce their alcohol intake to fewer than three units of alcohol. One unit of alcohol is defined as 10 mL of pure alcohol. This amount could be found in 25 mL of spirits (40% alcohol by volume), one third to one half a pint of beer (5-6% alcohol by volume), or half a standard glass of wine (12% alcohol by volume).
- Smoking cessation: it is recommended that all men at risk of osteoporosis stop smoking.
The American Urological Association 2018 Amendment of the Castration-Resistant Prostate Cancer similarly endorses preventative therapy in the form of supplemental calcium and vitamin D in men with castration-resistant prostate cancer.4 However, such interventions are likely advisable much earlier in the disease trajectory given the relatively minor risks of these supplements in contrast to the potentially debilitating and costly consequences of skeletal-related events. To this end, the NCCN and the National Osteoporosis Foundation recommend these interventions in all men over the age of 50 years who are receiving androgen deprivation therapy.
Pharmacologic interventions
In addition to calcium and vitamin D as highlighted above, the American Urological Association 2018 Amendment of the Castration-Resistant Prostate Cancer endorses pharmacotherapy with denosumab or zoledronic acid in men with castration-resistant prostate cancer.4
While further indications exist, the Endocrine Society Clinical Practice Guideline on Osteoporosis in Men recommends pharmacologic therapy in men at high risk for fracture based on, but not limited to, the following factors:3
- Men with a history of previous non-traumatic hip or vertebral fracture.
- Men with bone mineral density (T-score) that is 2.5 standard deviations or more below the mean of normal young white men, in the absence of spine or hip fractures.
- Men with a bone mineral density (T-score) from 1.0 to 2.5 standard deviations who also have an increased risk of fracture. In the United States, the suggested criteria for increased risk of fracture are a 10-year risk of any fracture equal to or exceeding 20% or a 10-year risk of hip fracture equal to or exceeding 3%, as determined using the FRAX tool. In other regions, the Guidelines recommend the utilization of region-specific guidelines.
- Men who are receiving long-term glucocorticoid treatment in significant doses defined by the 2010 American Society of Rheumatology as > 7.5 ng per day of prednisone or equivalent.
Pharmaceuticals - bisphosphonates
Bisphosphonates were among the first agents successfully used to prevent skeletal-related events in advanced prostate cancer. They function by reducing bone resorption through a variety of mechanisms including decreased osteoclast differentiation and survival and increased osteoblast survival. To do so, they compete with pyrophosphate for hydroxyapatite crystal binding sites, thus reducing osteoclast adherence to the bone. While there are a number of bisphosphonates used for a variety of clinical indications, zoledronic acid is most commonly used in patients with metastatic prostate cancer due to the Zometa 039 trial which demonstrated a reduction in SREs for patients who received zoledronic acid as compared to placebo.5 Based on these data, zoledronic acid is the only bisphosphonate approved for the prevention of skeletal-related events in men with metastatic prostate cancer.
Pharmaceuticals - denosumab
Denosumab is another agent which targets osteoclast activity. Bone exists in homeostasis between formation (driven by osteoblasts) and resorption (driven by osteoclasts). This homeostasis is regulated by the RANK (receptor activator of nuclear factor κB)/RANK-ligand system. Denosumab is a fully human monoclonal antibody that targets the RANK-ligand by mimicking OPG. Denosumab was first examined in postmenopausal women to prevent the development of osteoporosis and reduce the risk of fracture. Subsequently, denosumab was examined in women with breast cancer. Most relevantly, the Denosumab Protocol 20050103 compared denosumab and zoledronic acid in 1901 men with castrate-resistant prostate cancer.6 After a median follow-up of approximately one year, patients receiving denosumab had a significantly prolonged time to first-SRE (3.6 months incremental benefit, hazard ratio 0.82, 95% confidence interval 0.71 to 0.95). As a result of these data, denosumab was approved for the prevention of SREs in patients with metastatic solid tumors.
Pharmaceuticals – radio-isotopes
In addition to these osteoclast targeting agents, radio-isotopes may be used in the prevention of skeletal-related events. While beta-emitting particles (strontium-89 and samarium-153) may be used in the palliation of disease-related bony pain, they don’t have proven benefit in the prevention of SREs. In contrast, the alpha-emitting particle radium-223 has proven both palliative benefit as well as improvements in time to first SRE and overall survival. In the pivotal ALSYMPCA trial, 922 men with castrate-resistant prostate cancer and at least two symptomatic bony metastases who had either previously received or were unfit to receive docetaxel were randomized to radium-223 or placebo. Patients receiving radium-223 had significant improvements in time to first SRE (incremental benefit 5.2 months) as well as overall survival (incremental benefit in median survival of 2.8 months).2
Approach to preventing skeletal-related events
Philosophically, there are a number of times in the natural history of prostate cancer where a clinician may intervene to reduce the risk of prostate cancer related skeletal-related events. These relate to the complex interplay between prostate cancer and bone disease.
First, treatment may be directed at reducing or preventing fragility fractures due to prostate cancer-related therapy. In this disease space, lifestyle and nutrition-based interventions are paramount and are recommended by AUA guidelines as well as the National Comprehensive Cancer Network (NCCN). Pharmacologic interventions, including the use of osteoclast targeting therapies (bisphosphonates and RANK-ligand inhibitors), should be considered in this setting for patients at increased risk of fragility related fracture. Denosumab is FDA approved for this indication on the basis of the HALT 138 trial which found an increase in bone mineral density and a decrease in vertebral fractures among men receiving ADT for non-metastatic hormone-sensitive prostate cancer who receiving denosumab versus placebo.
Second, treatment may be directed at preventing bone metastases. In this disease space, bone-targeting agents including bisphosphonates and denosumab have been examined. The MRC PR04 trial demonstrated no benefit to clodronate in metastasis-free survival. Unfortunately, the Zometa 704 trial assessing the role of zoledronic acid in men with non-metastatic castration-resistant prostate cancer failed to accrue. However, the ZEUS trial accrued 1393 men with high-risk localized prostate cancer. Treatment with zoledronic acid failed to demonstrate an improvement in bone metastasis compared to placebo.7 The Denosumab Protocol 20050147 randomized men with high-risk nonmetastatic castrate-resistant prostate cancer to denosumab or placebo. Men receiving denosumab had prolonged metastasis-free survival (incremental benefit of 4.2 months).8
However, other approaches utilizing the suppression of the androgen axis in patients with non-metastatic castration-resistant prostate cancer (enzalutamide, apalutamide, and darolutamide) have demonstrated improvements in metastasis-free survival. While not a primary endpoint of these studies, it would be anticipated that delaying metastasis may improve skeletal-related events in these patients.
Third, we may seek to prevent skeletal-related events in men with known metastatic prostate cancer. This applies whether these men are in the castrate-sensitive or castrate-resistant disease space and it is here that the bulk of the evidence for prevention of SREs lies. In men with castrate-sensitive disease, CALGB 90202 demonstrated no improvement in skeletal-related events with the administration of zoledronic acid while castrate-sensitive rather than delayed initiation at the time of castration resistance.9 In contrast, there is significant evidence for the role of bone targeting agents in men with castrate-resistant disease. As previously mentioned, Zometa 039 demonstrated improvements in SREs for men receiving zoledronic acid compared with a placebo. Interestingly, CGP 032 and INT 05 failed to demonstrate a benefit to pamidronate. Thus, zoledronic acid is the only bisphosphonate approved in this space. The Denosumab Protocol 20050103 demonstrated an improvement in time to first SRE, as previously mentioned. Further, radio-isotope therapy using radium-223 was demonstrated in ALSYMPCA to improve time to first SRE in men with castrate-resistant prostate cancer and at least two symptomatic bony metastases.
Written by: Zachary Klaassen, MD, MSc, Assistant Professor of Urology, Georgia Cancer Center, Augusta University/Medical College of Georgia, Twitter: @zklaassen_md
Published Date: February 2020
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Morgans, Alicia K., and Matthew R. Smith. "Bone-targeted agents: preventing skeletal complications in prostate cancer." Urologic Clinics 39, no. 4 (2012): 533-546.
2. Parker, Christopher, S. Nilsson, Daniel Heinrich, Svein I. Helle, J. M. O'sullivan, Sophie D. Fosså, Aleš Chodacki et al. "Alpha emitter radium-223 and survival in metastatic prostate cancer." New England Journal of Medicine 369, no. 3 (2013): 213-223.
3. Watts, Nelson B., Robert A. Adler, John P. Bilezikian, Matthew T. Drake, Richard Eastell, Eric S. Orwoll, and Joel S. Finkelstein. "Osteoporosis in men: an Endocrine Society clinical practice guideline." The Journal of Clinical Endocrinology & Metabolism 97, no. 6 (2012): 1802-1822.
4. Lowrance, William T., Mohammad Hassan Murad, William K. Oh, David F. Jarrard, Matthew J. Resnick, and Michael S. Cookson. "Castration-resistant prostate cancer: AUA Guideline Amendment 2018." The Journal of urology 200, no. 6 (2018): 1264-1272.
5. Saad, Fred, Donald M. Gleason, Robin Murray, Simon Tchekmedyian, Peter Venner, Louis Lacombe, Joseph L. Chin, Jeferson J. Vinholes, J. Allen Goas, and Bee Chen. "A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma." Journal of the National Cancer Institute 94, no. 19 (2002): 1458-1468.
6. Fizazi, Karim, Michael Carducci, Matthew Smith, Ronaldo Damião, Janet Brown, Lawrence Karsh, Piotr Milecki et al. "Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study." The Lancet 377, no. 9768 (2011): 813-822.
7. Wirth, Manfred, Teuvo Tammela, Virgilio Cicalese, Francisco Gomez Veiga, Karl Delaere, Kurt Miller, Andrea Tubaro et al. "Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: efficacy and safety results of the Zometa European Study (ZEUS)." European urology 67, no. 3 (2015): 482-491
8. Smith, Matthew R., Fred Saad, Robert Coleman, Neal Shore, Karim Fizazi, Bertrand Tombal, Kurt Miller et al. "Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial." The Lancet 379, no. 9810 (2012): 39-46.
9. Smith, Matthew R., Susan Halabi, Charles J. Ryan, Arif Hussain, Nicholas Vogelzang, Walter Stadler, Ralph J. Hauke et al. "Randomized controlled trial of early zoledronic acid in men with castration-sensitive prostate cancer and bone metastases: results of CALGB 90202 (alliance)." Journal of Clinical Oncology 32, no. 11 (2014): 1143.
Radiopharmaceuticals in Prostate Cancer: Systemic “Bone-Seeking” Agents
Radiopharmaceuticals are pharmaceutical agents containing radioisotopes and emitting radiation that may be used for diagnostic or treatment purposes.
A number of small molecules have been used in conjunction with positron emission tomography (PET) scanning for prostate cancer staging. A recent presentation reported on the role of radiopharmaceutical driven imaging, predominately using Ga-PSMA, from the 2018 American Society of Clinical Oncology Annual Meeting.
From a therapeutic perspective, as they are typically given via intravenous infusion, radiopharmaceuticals are systemic radiotherapies, emitting alpha or beta radiation. Radiopharmaceuticals are indicated in patients with castrate-resistant prostate cancer with symptomatic bone metastases. Historically, beta-particle emitting agents including strontium-89 (Metastron®), samarium-153 (Quadramet®), phosphorus-32, and rhenium-186 were used as palliative therapies for patients with symptomatic bone disease.1 In this context, they are quite effective in relieving bony pain,2 however, these agents did not significantly improve survival.3 In contrast, the ALSYMPCA trial, which will be discussed in more detail below, demonstrated an improvement in both overall survival and skeletal-related events for patients receiving the alpha-emitter radium-223.4
In December 2018, the European Association of Nuclear Medicine Focus 1 Meeting reported a consensus regarding the use of molecular imaging and theranostics in prostate cancer.5 A number of relevant conclusions were derived following a systematic review and modified Delphi process. First, traditional diphosphate bone scan and contrast-enhanced computed tomography scan are mentioned but rarely recommended in the majority of patients in clinical guidelines. Second, magnetic resonance imaging and prostate cancer-targeted positron emission tomography are frequently suggested but the specific clinical scenarios in which they are most useful are poorly defined and how they may affect practice are poorly delineated. Third, sodium fluoride-18 positron emission tomography-CT bone scanning is not widely recommended; however, gallium-68 or fluorine-18 PSMA have gained acceptance. Finally, the palliative use of bone-targeting radiopharmaceuticals strontium-89, samarium-153, and rhenium-186 has been supplanted by radium-223, as well as other systemic therapies including docetaxel, abiraterone acetate, enzalutamide, and cabazitaxel.
Radium-223
Radium-223 dichloride (Xofigo®), commonly referred to as radium-223, is a targeted alpha emitter. It functions as a calcium mimetic and selectively binds newly forming bone stroma in regions of high bone turnover in osteoblastic or sclerotic bone metastasis.6 It then emits high-energy alpha particles with a very short range (less than 100 μm).7 This high-energy radiation induces a highly localized cytotoxic effect due to double-stranded DNA breakage.Initial Phase I and Phase II studies in patients with bone metastasis demonstrated radium-223 to be well tolerated, with minimal myelosuppression.8,9 Phase II trials also demonstrated that radium-223 effectively reduced bone-related pain and improved disease-related biomarkers, including bone alkaline phosphatase and prostate-specific antigen (PSA).9
As a result, the Phase III, Alpharadin in Symptomatic Prostate Cancer Patients (ALSYMPCA) trial was undertaken to assess the efficacy of radium-223 versus placebo in patients with metastatic castration-resistant prostate cancer (mCRPC) and bone metastases across 136 study centers in 19 countries.4 The trial enrolled patients with two or more bone metastases, detected on skeletal scintigraphy, without visceral metastasis who had previously received docetaxel, were docetaxel ineligible or declined docetaxel. Patients were required to have symptomatic disease, based on the requirement for regular analgesics or prior treatment with external beam radiotherapy for cancer-related bone pain in the preceding 12 weeks. Additionally, patients had to have a baseline PSA of at least 5 ng/mL with at least two progressive PSA rises; an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2; life expectancy of at least six months; and adequate hematologic, renal and liver function. Patients who had received chemotherapy within the preceding four weeks were excluded.
A total of 921 patients were enrolled and were randomized in a 2:1 ratio to radium-223 (50 kBq per kilogram of body weight intravenously), once every four weeks, plus best standard of care or placebo plus best standard of care.
The primary study endpoint was overall survival and secondary outcomes included time to first symptomatic skeletal event; biochemical endpoints including time to increase in total alkaline phosphatase level, total alkaline phosphatase response, normalization of alkaline phosphatase, time to increase in PSA; safety endpoints; and quality of life.
An initial, pre-specified, interim analysis was undertaken when 314 deaths had occurred. This demonstrated longer median overall survival among patients who received radium-223 (14.0 months) than those receiving placebo (11.2 months) with a resulting 30% decrease in the risk of death (hazard ratio 0.70, 95% 0.55 to 0.88). A subsequent, updated analysis was performed following 528 deaths. This demonstrated consistent results with longer median overall survival among patients who received radium-223 (14.9 months) than those receiving placebo (11.3 months). Similarly, the updated analysis confirmed a 30% reduction in the risk of death (hazard ratio [HR] 0.70, 95% confidence interval [CI] 0.58 to 0.83) for patients receiving radium-223. This benefit was observed across subgroups including total alkaline phosphatase level at randomization, current bisphosphonate use, previous docetaxel treatment, baseline ECOG score (0/1 vs 2), extent of disease (<6 metastases, 6-20 metastases, >20 metastases, and super scan), and opioid use.
Assessment of the secondary endpoints demonstrated a consistent benefit for radium-223. Notably, radium-223 delayed time to first symptomatic skeletal event (median, 15.6 months vs 9.8 months; HR 0.66, 95% CI 0.52 to 0.83). Unlike many systemic therapies, patients who received radium-223 were less likely to experience adverse events than those who received placebo: all adverse events (93% vs 96%), grade 3 or 4 adverse events (56% vs 62%), serious adverse events (47% vs 60%), and treatment-discontinuation as a result of adverse events (16% vs 21%). Finally, patients who received radium-223 were significantly more likely to have an improvement in the quality of life compared to patients receiving a placebo (p=0.02).
The authors subsequently published a pre-planned analysis with stratification according to receipt of prior docetaxel.10 Radium-223 prolonged survival both in patients who had previously received docetaxel (HR 0.70, 95% CI 0.56 to 0.88) and those who had not previously received docetaxel (HR 0.69 (95% CI 0.52 to 0.92).
As both radium-223 and abiraterone acetate11,12 have demonstrated survival benefits in patients with metastatic castrate-resistant prostate cancer, there was interest in combining these two agents. The ERA 223 trial randomized 806 patients with chemotherapy-naïve, metastatic castrate-resistant prostate cancer with bone metastasis to radium-223 or placebo, in addition to abiraterone acetate. Symptomatic skeletal event-free survival was the primary outcome. Somewhat unexpectedly, the trial was unblinded prematurely as more fractures and deaths were identified in the radium-223 arm than among patients receiving placebo. Median skeletal event-free survival was 22.3 months (interquartile range 17.0 to 25.8 months) among patients receiving radium-223 and abiraterone acetate and 26.0 months (interquartile range 21.8 months to 28.3 months) in patients receiving placebo and abiraterone acetate (HR 1.12, 95% CI 0.92 to 1.37). Fractures were more common among patients receiving radium-223 and abiraterone acetate (29%) than those receiving placebo and abiraterone acetate (11%). Thus, the combination of radium-223 and abiraterone acetate is not recommended in combination, however other combinations of agents with radium-223 are currently being tested (ie. enzalutamide).
Role of Radiopharmaceuticals in the AUA Guideline
The American Urological Association Guideline on Castrate-Resistant Prostate Cancer (amended in 2018) defines a number of clinical scenarios in which radiopharmaceuticals may be considered.13
First, among patients with good performance status and have not yet received docetaxel but who are symptomatic (based on a definition requiring regular use of narcotic analgesics for pain that is attributable to documented metastasis), radium-223 may be offered to patients who have symptoms attributable to bony metastatic disease in the absence of visceral disease in addition to standard of care options including abiraterone acetate plus prednisone, enzalutamide, and docetaxel. Patients with symptomatic metastases who decline these standard therapies, alternative treatments including radionuclide therapy (such as strontium-89) may be offered.13
Second, for symptomatic patients with poor performance status who have not previously received docetaxel, there is a relative paucity of direct evidence to inform treatment choices as most patients with poor performance status are excluded from clinical trials. Based on extrapolation from studies in patients with better performance status, the guideline recommends considering aggressive prostate cancer treatment where the functional impairments resulting in poor performance status are directly attributable to prostate cancer. In cases where the poor performance status is related to bony metastatic disease, radium-223 is a recommended option.13
Third, among patients with metastatic castrate-resistant prostate cancer who have previously received docetaxel-based chemotherapy, radium-223 is one of four agents with a proven survival benefit, along with abiraterone acetate plus prednisone, enzalutamide, and cabazitaxel.
Fourth, and finally, in patients with advanced mCRPC who are symptomatic and have poor performance status following previous docetaxel chemotherapy, symptom management is strongly advocated in keeping with the American Society for Clinical Oncology’s guidance regarding the treatment of patients with advanced solid tumors. However, judicious use of radionuclide therapy, along with abiraterone acetate plus prednisone, enzalutamide, ketoconazole plus steroids, are offered within the AUA guidelines13 despite the lack of strong data to support the use of these agents in this patient population.
The Canadian Urologic Association Guidelines similarly recommend radium-223 in patients with metastatic castrate-resistant prostate cancer who have bone pain related to their metastases and no visceral disease.14
New directions
The ALSYMPCA trial was the first to demonstrate that radiopharmaceuticals could improve overall survival, in addition to skeletal-related events,4 in patients with metastatic castrate-resistant prostate cancer. On the basis of this observation, there is an ongoing effort to identify molecular targets for linkage to radiopharmaceuticals. Proposed targets have included prostate-specific membrane antigen (PSMA) and gastrin-releasing peptide receptors (GRPr),15 These have the advantage of targeting prostate cancer cells, rather than being inherently bone targeting as is the case for current radiopharmaceuticals. Thus far, preliminary data based on prostate-specific membrane antigen targeted beta-emitters such as lutetium-177 suggest a promise to this approach but further work remains prior to the adoption of this approach.1 Further, ongoing research assessing prostate-specific membrane antigen targeted alpha-emitters is ongoing.
Written by: Zachary Klaassen, MD, MSc, Assistant Professor of Urology, Georgia Cancer Center, Augusta University/Medical College of Georgia, Atlanta, Georgia
Published Date: December 2019
- Written by: Christopher J.D. Wallis, MD, PhD and Zachary Klaassen, MD, MSc
- References:
1. Sartor, Oliver. "Isotope therapy for castrate-resistant prostate cancer: unique sequencing and combinations." The Cancer Journal 22, no. 5 (2016): 342-346.
2. Ye, Xiaojuan, Da Sun, and Cen Lou. "Comparison of the efficacy of strontium-89 chloride in treating bone metastasis of lung, breast, and prostate cancers." Journal of cancer research and therapeutics 14, no. 8 (2018): 36.
3. James, Nicholas, Sarah Pirrie, Ann Pope, Darren Barton, Lazaros Andronis, Ilias Goranitis, Stuart Collins et al. "TRAPEZE: a randomised controlled trial of the clinical effectiveness and cost-effectiveness of chemotherapy with zoledronic acid, strontium-89, or both, in men with bony metastatic castration-refractory prostate cancer." Health Technology Assessment 20 (2016).
4. Parker, Christopher, S. Nilsson, Daniel Heinrich, Svein I. Helle, J. M. O'sullivan, Sophie D. Fosså, Aleš Chodacki et al. "Alpha emitter radium-223 and survival in metastatic prostate cancer." New England Journal of Medicine 369, no. 3 (2013): 213-223.
5. Fanti, Stefano, Silvia Minozzi, Gerald Antoch, Ian Banks, Alberto Briganti, Ignasi Carrio, Arturo Chiti et al. "Consensus on molecular imaging and theranostics in prostate cancer." The Lancet Oncology 19, no. 12 (2018): e696-e708.
6. Henriksen, Gjermund, Knut Breistøl, Øyvind S. Bruland, Øystein Fodstad, and Roy H. Larsen. "Significant antitumor effect from bone-seeking, α-particle-emitting 223Ra demonstrated in an experimental skeletal metastases model." Cancer research 62, no. 11 (2002): 3120-3125.
7. Bruland, Øyvind S., Sten Nilsson, Darrell R. Fisher, and Roy H. Larsen. "High-linear energy transfer irradiation targeted to skeletal metastases by the α-emitter 223Ra: adjuvant or alternative to conventional modalities?." Clinical cancer research 12, no. 20 (2006): 6250s-6257s.
8. Nilsson, Sten, Roy H. Larsen, Sophie D. Fosså, Lise Balteskard, Kari W. Borch, Jan-Erik Westlin, Gro Salberg, and Øyvind S. Bruland. "First clinical experience with α-emitting radium-223 in the treatment of skeletal metastases." Clinical cancer research 11, no. 12 (2005): 4451-4459.
9. Nilsson, Sten, Lars Franzén, Christopher Parker, Christopher Tyrrell, René Blom, Jan Tennvall, Bo Lennernäs et al. "Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study." The lancet oncology 8, no. 7 (2007): 587-594.
10. Hoskin, Peter, Oliver Sartor, Joe M. O'Sullivan, Dag Clement Johannessen, Svein I. Helle, John Logue, David Bottomley et al. "Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: a prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial." The Lancet Oncology 15, no. 12 (2014): 1397-1406.
11. Ryan, Charles J., Matthew R. Smith, Johann S. De Bono, Arturo Molina, Christopher J. Logothetis, Paul De Souza, Karim Fizazi et al. "Abiraterone in metastatic prostate cancer without previous chemotherapy." New England Journal of Medicine 368, no. 2 (2013): 138-148.
12. De Bono, Johann S., Christopher J. Logothetis, Arturo Molina, Karim Fizazi, Scott North, Luis Chu, Kim N. Chi et al. "Abiraterone and increased survival in metastatic prostate cancer." New England Journal of Medicine 364, no. 21 (2011): 1995-2005.
13. Lowrance, William T., Mohammad Hassan Murad, William K. Oh, David F. Jarrard, Matthew J. Resnick, and Michael S. Cookson. "Castration-resistant prostate cancer: AUA Guideline Amendment 2018." The Journal of urology 200, no. 6 (2018): 1264-1272.
14. Saad, Fred, Kim N. Chi, Antonio Finelli, Sebastien J. Hotte, Jonathan Izawa, Anil Kapoor, Wassim Kassouf et al. "The 2015 CUA-CUOG Guidelines for the management of castration-resistant prostate cancer (CRPC)." Canadian Urological Association Journal 9, no. 3-4 (2015): 90.
15. Maffioli, L., L. Florimonte, D. Costa, C. Correia, C. Grana, M. Luster, L. Bodei, and M. Chinol. "New radiopharmaceutical agents for the treatment of castration-resistant prostate cancer." Quart J Nuclear Med Molec Imaging 59 (2015): 420-438.
Expanding Treatment Options in Non-metastatic Castrate-resistant Prostate Cancer
- Written by: Hanan Goldberg MD, Department of Urology, SUNY Upstate Medical University, Syracuse, NY, USA
- References:
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. Jan 2018;68(1):7-30.
- Trapasso JG, deKernion JB, Smith RB, Dorey F. The incidence and significance of detectable levels of serum prostate specific antigen after radical prostatectomy. J Urol. Nov 1994;152(5 Pt 2):1821-1825.
- Tombal B, Miller K, Boccon-Gibod L, et al. Additional analysis of the secondary end point of biochemical recurrence rate in a phase 3 trial (CS21) comparing degarelix 80 mg versus leuprolide in prostate cancer patients segmented by baseline characteristics. Eur Urol. May 2010;57(5):836-842.
- Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. Mar 2015;67(3):470-479.
- Saad F, Hotte SJ. Guidelines for the management of castrate-resistant prostate cancer. Canadian Urological Association journal = Journal de l'Association des urologues du Canada. 2010;4(6):380-384.
- Alpajaro SIR, Harris JAK, Evans CP. Non-metastatic castration resistant prostate cancer: a review of current and emerging medical therapies. Prostate Cancer Prostatic Dis. Mar 2019;22(1):16-23.
- Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. Jun 2015;4(3):365-380.
- Sharifi N, Dahut WL, Steinberg SM, et al. A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU Int. Nov 2005;96(7):985-989.
- Macomson B, Lin JH, Tunceli O, et al. Time to metastasis or death in non-metastatic castrate resistant prostate cancer (nmCRPC) patients by National Comprehensive Cancer Network (NCCN) risk groups. Journal of Clinical Oncology. 2017;35(15_suppl):5027-5027.
- Scher HI, Solo K, Valant J, Todd MB, Mehra M. Prevalence of Prostate Cancer Clinical States and Mortality in the United States: Estimates Using a Dynamic Progression Model. PLoS One. 2015;10(10):e0139440.
- Lowrance WT, Murad MH, Oh WK, Jarrard DF, Resnick MJ, Cookson MS. Castration-Resistant Prostate Cancer: AUA Guideline Amendment 2018. J Urol. Dec 2018;200(6):1264-1272.
- Scher HI, Morris MJ, Stadler WM, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. Apr 20 2016;34(12):1402-1418.
- Liede A, Arellano J, Hechmati G, Bennett B, Wong S. International prevalence of nonmetastatic (M0) castration-resistant prostate cancer (CRPC). Journal of Clinical Oncology. 2013;31(15_suppl):e16052-e16052.
- Smith MR, Saad F, Chowdhury S, et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. New England Journal of Medicine. 2018;378(15):1408-1418.
- Virgo KS, Rumble RB, Singer EA. Second-Line Hormonal Therapy for Men With Chemotherapy-Naive Castration-Resistant Prostate Cancer: American Society of Clinical Oncology Provisional Clinical Opinion Summary. J Oncol Pract. Jul 2017;13(7):459-461.
- Crawford ED, Stone NN, Yu EY, et al. Challenges and recommendations for early identification of metastatic disease in prostate cancer. Urology. Mar 2014;83(3):664-669.
- Sartor AO, Tangen CM, Hussain MH, et al. Antiandrogen withdrawal in castrate-refractory prostate cancer: a Southwest Oncology Group trial (SWOG 9426). Cancer. Jun 2008;112(11):2393-2400.
- Murray NP, Reyes E, Tapia P, Badínez L, Orellana N. Differential expression of matrix metalloproteinase-2 expression in disseminated tumor cells and micrometastasis in bone marrow of patients with nonmetastatic and metastatic prostate cancer: theoretical considerations and clinical implications-an immunocytochemical study. Bone marrow research. 2012;2012:259351-259351.
- Eiber M, Maurer T, Souvatzoglou M, et al. Evaluation of Hybrid (6)(8)Ga-PSMA Ligand PET/CT in 248 Patients with Biochemical Recurrence After Radical Prostatectomy. J Nucl Med. May 2015;56(5):668-674.
- Morigi JJ, Stricker PD, van Leeuwen PJ, et al. Prospective Comparison of 18F-Fluoromethylcholine Versus 68Ga-PSMA PET/CT in Prostate Cancer Patients Who Have Rising PSA After Curative Treatment and Are Being Considered for Targeted Therapy. J Nucl Med. Aug 2015;56(8):1185-1190.
- Umbehr MH, Muntener M, Hany T, Sulser T, Bachmann LM. The role of 11C-choline and 18F-fluorocholine positron emission tomography (PET) and PET/CT in prostate cancer: a systematic review and meta-analysis. Eur Urol. Jul 2013;64(1):106-117.
- Geynisman DM, Plimack ER, Zibelman M. Second-generation Androgen Receptor-targeted Therapies in Nonmetastatic Castration-resistant Prostate Cancer: Effective Early Intervention or Intervening Too Early? Eur Urol. Dec 2016;70(6):971-973.
- Taylor CD, Elson P, Trump DL. Importance of continued testicular suppression in hormone-refractory prostate cancer. J Clin Oncol. Nov 1993;11(11):2167-2172.
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. Nov 10 2005;23(32):8253-8261.
- Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. Dec 15 2004;64(24):9209-9216.
- Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists' Collaborative Group. Lancet. Apr 29 2000;355(9214):1491-1498.
- Schweizer MT, Zhou XC, Wang H, et al. Metastasis-free survival is associated with overall survival in men with PSA-recurrent prostate cancer treated with deferred androgen deprivation therapy. Ann Oncol. Nov 2013;24(11):2881-2886.
- Schroder FH, Tombal B, Miller K, et al. Changes in alkaline phosphatase levels in patients with prostate cancer receiving degarelix or leuprolide: results from a 12-month, comparative, phase III study. BJU Int. Jul 2010;106(2):182-187.
- Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. May 8 2009;324(5928):787-790.
- Hussain M, Fizazi K, Saad F, et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. New England Journal of Medicine. 2018;378(26):2465-2474.
- Fizazi K, Shore N, Tammela TL, et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. New England Journal of Medicine. 2019;380(13):1235-1246.
- El-Amm J, Aragon-Ching JB. The Current Landscape of Treatment in Non-Metastatic Castration-Resistant Prostate Cancer. 2019;13:1179554919833927.
- Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. Mar 15 2012;72(6):1494-1503.
- Scher HI, Fizazi K, Saad F, et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. New England Journal of Medicine. 2012;367(13):1187-1197.
- Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. Jul 31 2014;371(5):424-433.
- Xie W, Regan MM, Buyse M, et al. Metastasis-Free Survival Is a Strong Surrogate of Overall Survival in Localized Prostate Cancer. J Clin Oncol. Sep 20 2017;35(27):3097-3104.
- Shore ND. Darolutamide (ODM-201) for the treatment of prostate cancer. Expert Opin Pharmacother. Jun 2017;18(9):945-952.
- Smith MR, Saad F, Oudard S, et al. Denosumab and bone metastasis-free survival in men with nonmetastatic castration-resistant prostate cancer: exploratory analyses by baseline prostate-specific antigen doubling time. J Clin Oncol. Oct 20 2013;31(30):3800-3806.
- Nelson JB, Love W, Chin JL, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer. Nov 1 2008;113(9):2478-2487.
- Miller K, Moul JW, Gleave M, et al. Phase III, randomized, placebo-controlled study of once-daily oral zibotentan (ZD4054) in patients with non-metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. Jun 2013;16(2):187-192.
- Beinart G, Rini BI, Weinberg V, Small EJ. Antigen-presenting cells 8015 (Provenge) in patients with androgen-dependent, biochemically relapsed prostate cancer. Clin Prostate Cancer. Jun 2005;4(1):55-60.
- Madan RA, Gulley JL, Schlom J, et al. Analysis of overall survival in patients with nonmetastatic castration-resistant prostate cancer treated with vaccine, nilutamide, and combination therapy. Clin Cancer Res. Jul 15 2008;14(14):4526-4531.
- Ogita S, Tejwani S, Heilbrun L, et al. Pilot Phase II Trial of Bevacizumab Monotherapy in Nonmetastatic Castrate-Resistant Prostate Cancer. ISRN oncology. 2012;2012:242850-242850.
- Ryan CJ, Crawford ED, Shore ND, et al. The IMAAGEN Study: Effect of Abiraterone Acetate and Prednisone on Prostate Specific Antigen and Radiographic Disease Progression in Patients with Nonmetastatic Castration Resistant Prostate Cancer. J Urol. Aug 2018;200(2):344-352.
- Hussain M, Corn PG, Michaelson MD, et al. Phase II study of single-agent orteronel (TAK-700) in patients with nonmetastatic castration-resistant prostate cancer and rising prostate-specific antigen. Clin Cancer Res. Aug 15 2014;20(16):4218-4227.
- Fizazi K, Jones R, Oudard S, et al. Phase III, randomized, double-blind, multicenter trial comparing orteronel (TAK-700) plus prednisone with placebo plus prednisone in patients with metastatic castration-resistant prostate cancer that has progressed during or after docetaxel-based therapy: ELM-PC 5. J Clin Oncol. Mar 1 2015;33(7):723-731.
The Current Status of Stereotactic Body Radiation Therapy in Kidney Cancer
There are a number of accepted treatment options for patients diagnosed with localized RCC. These include radical nephrectomy (whether open, laparoscopic or robotic), partial nephrectomy (whether open, laparoscopic, or robotic), surgical or non-surgical ablation, and active surveillance. The most appropriate treatment strategy will depend on patient (host) and tumor characteristics. These details are discussed more fully in the “Malignant Renal Tumors” article in the UroToday Center of Excellence series.
Kidney cancer has been historically thought of as a “radio-resistant” tumor. This is based on in vitro studies2 as well as the fact that early trial of adjuvant and neoadjuvant radiotherapy in patients with RCC undergoing surgical resection failed to show benefit.3,4 As a result, traditionally fractionated radiotherapy has been historically limited to palliative intent for patients with RCC. However, hypofractionated, high-dose radiotherapy has proven successful in the local control of RCC metastasis to the brain and other bony and visceral sites (refs 6-15). Coinciding with these clinical data was the emergence of data demonstrating the efficacy of high dose per fraction radiotherapy in the treatment of RCC in a mouse model.5 This led to increasing interest in the use of stereotactic body radiotherapy (SBRT) in the treatment of localized RCC. SBRT is routinely used for the treatment of malignancies of other tissue types including lung, liver, spine, and prostate.6 Compared to other radiation techniques, SBRT utilizes a smaller number of higher dose fractions. This is believed to assist with overcoming the previously believed radioresistance of RCC. Further, compared to other ablative approaches, one of the advantages of SBRT is the ability to treat larger lesions.6
Given uncertainties about both the efficacy and toxicity of such an approach, initial investigation has focused on patients in whom extirpative surgery, the gold standard approach, is not feasible or safe.
There are currently both retrospective and prospective reports characterising outcomes for patients treated with SBRT for localized RCC. These studies include a variety of treatment approaches including single fraction treatment (often 26 Gy in 1 fraction) and multiple fraction regimes (including regimes ranging from 2 to 10 fractions and total doses ranging from 5 to 85 Gy). As may be expected from some different treatment approaches, there are differences in both efficacy and toxicity between studies.
Prospective cohort studies
A recent systematic review identified eight published prospective studies of SBRT in the treatment of patients with localized RCC.7 Apart from one study published in 2006, the remainder have been published in the last five years. The strength of conclusions that can be drawn from these data are limited by small sample sizes (4 to 40 patients with localized RCC per study) and limited follow-up (13 to 52 months, with most 2 years or less).7 In addition, as previously mentioned, there were significant differences in total dose delivered and radiotherapy prescription between studies.In each case, the authors report on patients who were either deemed medically inoperable, at very high risk for surgery due to the risk of dialysis or who refused surgery. Some studies had specific, disease-related criteria (e.g. a single lesion, maximal tumor dimension less than 4 or 5 cm) whereas this was not specified in other manuscripts. Outcomes were variably reported with local control most often reported. Additionally, adverse events were variously, and non-systematically reported.
Local control rates varied, in large part in correlation to the duration of observation: from 87% local control rate at a median 37 months follow up to 100% at two years in one trial8. Notably, even in the publication from Siva and colleagues who reported 100% local control, 10% of patients experienced distant progression, an outcome that is more likely to contribute to morbidity and mortality than local recurrence.8 Longer-term outcomes remain to be assessed.
Likely due in part to differing radiotherapy prescriptions, toxicity rates varied significantly. Both Siva and colleagues and Svendman and colleagues reported grade 1-2 toxicity in more than 50% of patients, most notably characterized by chest wall pain, nausea, and fatigue.8,9 In addition to considerations regarding comorbidity, SBRT and other non-surgical approaches to renal masses are often considered in patients with poor renal function for whom nephron preservation is a top priority. Thus, post-procedural renal function is an important outcome and, again, results vary between reports. Kaplan and colleagues reported worsening of renal function in 2 of 12 patients undergoing SBRT for medically inoperable tumors less than 5cm.10 McBride et al., in a similar population of patients, found that 2 of 15 patients (13%) had late grade 3 renal dysfunction with a mean decrease in glomerular filtration rate of 18 mg/dL among the whole study population.11 Similarly, Ponsky and colleagues demonstrated an 11% rate of grade 3 renal dysfunction among 19 patients deemed poor surgical candidates who received SBRT.12 Finally, and perhaps more optimistically, Siva and colleagues found in their cohort of 21 patients that the average decrease in glomerular filtration rate was only 8.7 mL/min at one year following treatment.8. Taken together, evidence suggests that increased fractionation (as in 20 to 30 Gy in 10 fractions) was strongly correlated with renal atrophy.13
Taken together, these data suggest that, for patients who receive three radiation fractions, a minimum per fraction dose of 11 Gy should be administered as this was the minimum dose that, in prospective cohorts, no patient experienced local failure.7
Retrospective cohort studies
In addition to the aforementioned prospective cohort studies, there are a number of retrospective cohort studies examining the use of SBRT in primary RCC. These, for the most part, have the same limitations are the prospective studies including limited sample size, short follow-up and heterogeneity of radiotherapy prescription. While most of these reports demonstrated local control rates comparable to the prospective literature (93 – 100%), one study demonstrated significantly lower local control (65%) among patients who had a history of radical nephrectomy for RCC in the contra-lateral kidney14. Those patients received 60 to 85 Gy in 5 to 7 fractions using stereotactic gamma-ray irradiation.
Patient selection
Surgery remains the mainstay of curative-intent treatment for patients with localized RCC. Ablative approaches, including SBRT, may, therefore, be considered among patients for whom surgery is contraindicated or who refuse surgery. Recent guidelines have recommended emphasizing that SBRT remains an experimental option in RCC due to the relatively limited worldwide experience and lack of long-term data.15Treatment recommendations
The International Radiosurgery Oncology Consortium for Kidney performed a 65 item survey among eight institutions who performed SBRT for primary RCC.16 A number of important conclusions came out of this work and the resulting consensus statement. First, all included centers treat patients with solitary kidneys or pre-existing hypertension. Five of the eight institutions have size cut off criteria ranging from 5 to 8 cm in maximal tumor dimension. The total planning target volume expansion varied between institutions, ranging from 3 to 10 mm. While all centers used pretreatment image verification, seven of the eight utilized intrafractional monitoring of some sort. Radiation prescriptions varied from 1 to 12 fractions with a total dose of 25 to 80 Gy. However, the consensus statement recommends a total dose of 36 to 45 Gy for patients receiving 3 fraction regimes and 40 to 50 Gy for patients receiving 5 fraction regimes. Obviously, the size of the primary tumour and its proximity to critical and adjacent structures will influence the total dose and fractionation regime.Ongoing surveillance follow-up for local tumor response and recurrence varied with some institutions relying on computed tomography (CT) alone while others used magnetic resonance imaging (MRI) or PET-CT. Typically, follow-up was performed every three to six months in the first two years and every three to twelve months in the subsequent three years.
One of the challenges in the post-treatment monitoring of these patients is the interpretation of radiographic studies and identification of imaging studies. Among 41 tumours treated with SBRT, the largest available study of imaging characteristics following treatment found that the linear growth rate regressed by an average of 0.37 cm per year after treatment but that there were no significant changed in enhancement when comparing imaging before and following treatment.17
Conclusions
Stereotactic body radiotherapy is an emerging treatment approach for patients with primary renal cell carcinoma. Compared to other ablative approaches (such as radiofrequency ablation or cryotherapy), it offers the opportunity to treat larger tumors and potentially those in closer proximity to critical structures. To date, surgical extirpation via partial or radical nephrectomy remains the gold standard and SBRT has primarily been investigated among patients who are either deemed medically inoperable or who refuse surgery. There are a number of ongoing studies assessing the role of SBRT that will increase the prospective global experience with this approach, however, none will provide comparative data with other treatment approaches. One particular area of interest in the potential for synergistic effects between SBRT and systemic therapy, particularly immunotherapy (ClinicalTrials.gov identifiers: NCT01896271, NCT02781506, NCT02306954, and NCT02334709).
Published Date: November 2019
- Written by: Zachary Klaassen, MD MSc
- References:
References:
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68(1):7-30.
2. Deschavanne PJ, Fertil B. A review of human cell radiosensitivity in vitro. Int J Radiat Oncol Biol Phys 1996; 34(1):251-66.
3. Finney R. The value of radiotherapy in the treatment of hypernephroma--a clinical trial. Br J Urol 1973; 45(3):258-69.
4. Juusela H, Malmio K, Alfthan O, et al. Preoperative irradiation in the treatment of renal adenocarcinoma. Scand J Urol Nephrol 1977; 11(3):277-81.
5. Walsh L, Stanfield JL, Cho LC, et al. Efficacy of ablative high-dose-per-fraction radiation for implanted human renal cell cancer in a nude mouse model. Eur Urol 2006; 50(4):795-800; discussion 800.
6. Francolini G, Detti B, Ingrosso G, et al. Stereotactic body radiation therapy (SBRT) on renal cell carcinoma, an overview of technical aspects, biological rationale and current literature. Crit Rev Oncol Hematol 2018; 131:24-29.
7. Miccio J, Johung K. When Surgery Is Not an Option in Renal Cell Carcinoma: The Evolving Role of Stereotactic Body Radiation Therapy. Oncology (Williston Park) 2019; 33(5):167-73, 177.
8. Siva S, Pham D, Kron T, et al. Stereotactic ablative body radiotherapy for inoperable primary kidney cancer: a prospective clinical trial. BJU Int 2017; 120(5):623-630.
9. Svedman C, Sandstrom P, Pisa P, et al. A prospective Phase II trial of using extracranial stereotactic radiotherapy in primary and metastatic renal cell carcinoma. Acta Oncol 2006; 45(7):870-5.
10. Kaplan ID, Redrosa I, C. M, et al. Results of a phase I dose escalation study of stereotactic radiosurgery for primary renal tumors. Int J Radiat Oncol Biol Phys 2010; 78:S191.
11. McBride SM, Wagner AA, Kaplan ID. A phase 1 dose-escalation study of robotic radiosurgery in inoperable primary renal cell carcinoma. Int J Radiat Oncol Biol Phys 2013; 87:S84.
12. Ponsky L, Lo SS, Zhang Y, et al. Phase I dose-escalation study of stereotactic body radiotherapy (SBRT) for poor surgical candidates with localized renal cell carcinoma. Radiother Oncol 2015; 117(1):183-7.
13. Yamamoto T, Kadoya N, Takeda K, et al. Renal atrophy after stereotactic body radiotherapy for renal cell carcinoma. Radiat Oncol 2016; 11:72.
14. Wang YJ, Han TT, Xue JX, et al. Stereotactic gamma-ray body radiation therapy for asynchronous bilateral renal cell carcinoma. Radiol Med 2014; 119(11):878-83.
15. Muller AC, van Oorschot B, Micke O, et al. [German S3 guideline for renal cell carcinoma : Presentation and discussion of essential aspects for the radiation oncologist]. Strahlenther Onkol 2018; 194(1):1-8.
16. Siva S, Ellis RJ, Ponsky L, et al. Consensus statement from the International Radiosurgery Oncology Consortium for Kidney for primary renal cell carcinoma. Future Oncol 2016; 12(5):637-45.
17. Sun MR, Brook A, Powell MF, et al. Effect of Stereotactic Body Radiotherapy on the Growth Kinetics and Enhancement Pattern of Primary Renal Tumors. AJR Am J Roentgenol 2016; 206(3):544-53.
Contemporary Radiotherapy Clinical Trials for Prostate Cancer
Localized Treatment – Optimizing Fractionation
Several trials have recently focused on delineating the optimal radiotherapy fractionation schedule for the primary treatment of prostate cancer. In 2016, efficacy results of the Dutch HYPRO trial were published, assessing hypofractionated radiotherapy compared with conventionally fractionated radiotherapy among patients with intermediate-risk to high-risk T1b-T4NX-N0MX-M0 localized prostate cancer.1 Patients were assigned 1:1 to either hypofractionated radiotherapy of 64.6 Gy (19 fractions of 3.4 Gy, three fractions per week) or conventionally fractionated radiotherapy of 78.0 Gy (39 fractions of 2.0 Gy, five fractions per week) with a primary endpoint of relapse-free survival. There were 804 patients assessed in the intention-to-treat analysis, of which 407 were assigned hypofractionated radiotherapy and 397 were allocated to conventionally fractionated radiotherapy. Additionally, 67% of patients received concomitant androgen deprivation therapy (ADT) for a median duration of 32 months (IQR 10-44). Over a median follow-up of 60 months (IQR 51-69), treatment failure was reported in 21% of patients, including 20% in the hypofractionation group and 22% in the conventional fractionation group. The 5-year relapse-free survival was 80.5% (95% CI 75.7-84.4) for patients assigned hypofractionation and 77.1% (71.9-81.5) for those allocated conventional fractionation (hazard ratio [HR] 0.86, 95% confidence interval [CI] 0.63-1.16; log-rank p=0.36). Based on these results, the authors noted that this current regimen of hypofractionated radiotherapy was not superior to conventional radiotherapy.
The CHHiP trial was a multi-center, randomized, Phase III trial, designed as a non-inferiority clinical trial, randomizing men with pT1b-T3aN0M0 prostate cancer 1:1:1 to conventional (74 Gy delivered in 37 fractions over 7.4 weeks) or one of two hypofractionated schedules (60 Gy in 20 fractions over 4 weeks or 57 Gy in 19 fractions over 3.8 weeks) all delivered with intensity-modulated techniques.2 Most patients were given radiotherapy with 3-6 months of neoadjuvant and concurrent androgen suppression. The primary endpoint was time to biochemical or clinical failure, and the critical hazard ratio for non-inferiority was 1.208. In this large trial, 3,216 men were enrolled from 71 centers and randomly assigned: 1,065 patients to the 74 Gy group, 1,074 patients to the 60 Gy group, and 1,077 patients to the 57 Gy group. The median follow-up was 62.4 months (IQR 53.9-77.0) over which the proportion of patients who were biochemical or clinical failure-free at five years was 88.3% (95% CI 86.0-90.2) in the 74 Gy group, 90.6% (95% CI 88.5-92.3) in the 60 Gy group, and 85.9% (95% CI 83.4-88.0) in the 57 Gy group. The 60 Gy hypofractionated schedule was non-inferior to the conventional 74 Gy schedule (HR 0.84, 90% CI 0.68-1.03, p noninferiority = 0.0018), but non-inferiority was not possible for 57 Gy hypofractionation compared with 74 Gy (HR 1.20, 90% CI 0.99-1.46, p noninferiority = 0.48).
Based on these initial studies, recent interest has related to even more intense radiotherapy fractionation. The Scandinavian HYPO-RT-PC randomized controlled Phase III trial was initially presented at ESTRO 2018, and subsequently published in Lancet Oncology.3 This trial randomized men with intermediate and high-risk prostate cancer to either conventional fractionating (n = 602; 78.0 Gy in 39 fractions, 5 days per week for 8 weeks) or ultrahypofractionated (n=598; 42.7 Gy in seven fractions, three days per week for 2.5 weeks). The primary endpoint was time to biochemical or clinical failure. The estimated failure-free survival at five years was 84% (95% CI 80-87) in both treatment groups, with an adjusted HR of 1.002 (95% CI 0.758-1.325; log-rank p=0.99). There was weak evidence of an increased frequency of acute physician-reported RTOG grade 2 or worse urinary toxicity in the ultra-hypofractionation group at end of radiotherapy (158 [28%] of 569 patients vs 132 [23%] of 578 patients; p=0.057). Based on these results, there has been support for the use of ultra-hypofractionated radiotherapy for prostate cancer.
However, despite encouraging results regarding biochemical/relapse-free survival as described above, studies have not demonstrated an overall survival (OS) benefit. Patients in the NRG Oncology RTOG 0126 trial that had intermediate-risk prostate cancer were randomized to 3-dimensional conformal radiation therapy or intensity-modulated radiation therapy to 79.2 Gy in 44 fractions or 70.2 Gy in 39 fractions.4 Over a median follow-up of 8.4 years in 1,499 patients, there was no difference in OS between the 751 men in the 79.2-Gy arm and the 748 men in the 70.2-Gy arm. The 8-year rates of OS were 76% with 79.2 Gy and 75% with 70.2 Gy (HR 1.00, 95% CI, 0.83-1.20), and the 8-year cumulative rates of distant metastases were 4% for the 79.2-Gy arm and 6% for the 70.2-Gy arm (HR 0.65, 95% CI, 0.42-1.01). As the above trials continue to mature, it will be imperative to evaluate survival outcomes. However, adoption of hypofractionation needn’t require improved survival or toxicity outcomes – on the basis of equivalence, improvements in the patient experience/convenience and cost would support the adoption of this approach.
Localized Disease – Optimizing Androgen Deprivation Therapy (ADT)
A second initiative of recent radiotherapy trials has been delineating the appropriate dose of ADT given concurrently with radiotherapy. The DART01/05 GICOR trial was a randomized, controlled Phase III trial assessing high-dose radiotherapy with short-term or long-term ADT for patients with T1c-T3b N0M0 prostate cancer with intermediate-risk and high-risk features.5 Patients were randomly assigned 1:1 to receive either four months of ADT combined with three-dimensional conformal radiotherapy at a minimum dose of 76 Gy (range 76-82 Gy; short-term ADT group) or the same treatment followed by 24 months of adjuvant ADT (long-term ADT group), stratified by prostate cancer risk group (intermediate risk vs high risk). In this Spanish multi-center trial, 178 patients were randomly assigned to receive short-term ADT and 177 to receive long-term ADT. After a median follow-up of 63 months (IQR 50-82), 5-year biochemical disease-free survival was significantly better among patients receiving long-term ADT than among those receiving short-term ADT: 90% (95% CI 87-92) vs 81% (95% CI 78-85), HR 1.88, 95% CI 1.12-3.15. Furthermore, the 5-year OS (95% vs 86%; HR 2.48, 95% CI 1.31-4.68) and 5-year MFS (94% vs 83%; HR 2.31, 95% CI 1.23-3.85) rates were also significantly better in the long-term ADT group than in the short-term ADT group. Not surprisingly, the authors noted that the effect of long-term ADT on biochemical disease-free survival, metastasis-free survival, and overall survival was more evident in patients with high-risk disease than in those with low-risk disease.
In 2016, the results of the EORTC 22991 randomized trial were published.6 This trial assessed if biochemical disease-free survival (DFS) is improved by adding six months of androgen suppression to primary radiotherapy for intermediate- or high-risk localized prostate cancer. Among 819 patients, the median patient age was 70 years, 74.8% were intermediate risk and 24.8% were high risk. At 7.2 years median follow-up, radiotherapy plus androgen suppression significantly improved biochemical DFS (HR 0.52, 95% CI 0.41-0.66), as well as clinical progression-free survival (HR 0.63, 95% CI 0.48-0.84).
Long-term follow-up of the D’Amico trial assessing ADT plus radiotherapy versus ADT alone for patients with localized, unfavorable risk prostate cancer was published in 20157. This analysis importantly showed that underlying comorbidity significantly modified the benefit of ADT: in patients with moderate or severe comorbidity, use of radiotherapy alone was associated with decreased cardiac mortality (HR 0.17, 95% CI 0.06-0.46), non-prostate cancer mortality (HR 2.79, 95% CI 1.02-7.60), and overall mortality (HR 0.36, 95% CI 0.19-0.67). Conversely, in men with no or minimal comorbidity, radiotherapy alone was associated with an increased risk of overall mortality and prostate cancer mortality, without coinciding decreases in cardiac mortality or non-prostate cancer mortality.
The prior standard ADT duration was 28-36 months when combined with radiotherapy for high-risk disease. As such, the TROG RADAR trial assessed whether the addition of 12 months of adjuvant ADT, 18 months of zoledronic acid, or both, can improve outcomes in men with locally advanced prostate cancer who receive six months of ADT and prostate radiotherapy.8 In 2019, this trial reported 10-year outcomes. There were 1,071 patients randomized to radiotherapy plus six months of ADT or radiotherapy plus 18 months of ADT. This trial found that 18 months of ADT plus radiotherapy is a more effective treatment option for locally advanced prostate cancer than six months of ADT plus radiotherapy (HR 0.70, 95% CI 0.50-0.98), but the addition of zoledronic acid to this treatment regimen is not beneficial. Finally, the PCS IV Trial randomized 630 patients with a median follow-up of 9.4 years to radiotherapy plus 18 months of ADT or radiotherapy plus 36 months of ADT.9 The 5-year OS rates were 91% for long term ADT arm (95% CI 88-95%) and 86% for short term ADT arm (95% CI 83-90%, p=0.07). The quality of life analysis showed a significant difference (p<0.001) in six scales and 13 items favoring 18 months of ADT.
Localized Disease – Addition of Chemotherapy
The benefit of adding chemotherapy in the treatment of very high-risk disease has also been recently evaluated. Rosenthal et al.10 published the multicenter randomized NRG Oncology RTOG 0521 clinical trial, which enrolled patients with high-risk nonmetastatic disease between 2005 and 2009. Patients were randomly assigned (n=563) to receive standard long-term ADT plus radiotherapy with or without adjuvant chemotherapy. Over a median follow-up of 5.7 years, the 4-year OS rate was 89% (95% CI, 84% to 92%) for ADT and radiotherapy, compared to 93% (95% CI, 90% to 96%) for ADT and radiotherapy plus chemotherapy (HR 0.69, 90% CI 0.49-0.97). Six-year rate of distant metastasis was 14% for ADT and radiotherapy and 9.1% for ADT and radiotherapy plus chemotherapy, (HR 0.60, 95% CI 0.37-0.99).
Treatment after Radical Prostatectomy
For decades, urologists and radiation oncologists have debated the optimal timing, location, and dose of radiotherapy, in addition to the utilization of ADT among those experiencing biochemical recurrence after radical prostatectomy. The much-anticipated NRG Oncology/RTOG 0534 SPPORT trial reported initial results at the 2019 ASTRO meeting.11 This trial randomized 1,736 patients to either (i) salvage radiotherapy to the prostate bed, (ii) salvage radiotherapy to the prostate bed plus ADT, or (iii) salvage radiotherapy to the prostate bed plus ADT plus radiotherapy to the pelvic lymph nodes. The 5-year freedom from progression rate was 71% for salvage radiotherapy to the prostate bed, 81% for salvage radiotherapy to the prostate bed plus ADT, and 87% for salvage radiotherapy to the prostate bed plus ADT plus radiotherapy to the pelvic lymph nodes. Based on only 108 patients with metastasis, there were minimal differences between the three arms with regards to metastasis-free survival. It is likely that with continued follow-up, these results will likely continue to favor salvage radiotherapy to the prostate bed plus ADT plus radiotherapy to the pelvic lymph nodes.
Previously, Shipley et al. assessed whether antiandrogen therapy with radiation therapy improves cancer control and prolong OS among patients with biochemical recurrence.12 In this trial, there were 760 patients who had undergone radical prostatectomy with lymphadenectomy and had biochemically recurrent disease who were randomized to radiation therapy and to receive either antiandrogen therapy (24 months of bicalutamide at a dose of 150 mg daily) or daily placebo tablets during and after radiation therapy. The primary endpoint was the OS rate; the actuarial rate of OS at 12 years was 76.3% in the bicalutamide group, as compared with 71.3% in the placebo group (HR 0.77, 95% CI 0.59 to 0.99). The 12-year incidence of death from prostate cancer was 5.8% in the bicalutamide group, as compared with 13.4% in the placebo group (p < 0.001). Finally, the cumulative incidence of metastatic prostate cancer at 12 years was 14.5% in the bicalutamide group, as compared with 23.0% in the placebo group (p = 0.005). As such, the addition of antiandrogen improved clinical outcomes and OS in patients with biochemical recurrence after radical prostatectomy.
Several randomized trials assessing adjuvant vs salvage radiotherapy have been presented earlier this year at academic conferences, but have yet to be published.
Radiotherapy in the Setting of Metastatic Disease
Several trials have recently assessed the impact of radiotherapy to the prostate in the setting of metastatic disease, given retrospective evidence that patients may derive a survival benefit for treatment to the primary prostate tumor. The HORRAD trial was a multi-center randomized controlled trial recruiting 432 patients with a prostate-specific antigen (PSA) >20ng/ml and primary bone metastatic prostate cancer on bone scan (between 2004 and 2014).13 Patients were randomized to either ADT with external beam radiotherapy or ADT alone. The median PSA level was 142ng/ml and 67% of patients had more than five osseous metastases. Over a median follow up of 47 months, the median OS was 45 months (95% CI, 40.4-49.6) in the radiotherapy group and 43 months (95% CI, 32.6-53.4) in the control group (p = 0.4; HR 0.90, 95% CI 0.70-1.14). The median time to PSA progression in the radiotherapy group was 15 months (95% CI, 11.8-18.2), compared with 12 months (95% CI, 10.6-13.4) in the control group.
Despite the negative results from HORRAD, there was much anticipation for the results of the STAMPEDE arm H clinical trial assessing the efficacy of radiotherapy to the primary in M1 disease.14 This study randomized 2,061 to either standard systemic treatments (ADT +/- chemotherapy) versus standard systemic treatments (ADT +/- chemotherapy) plus radiotherapy to the primary. There were 819 (40%) men that had a low metastatic burden, 1,120 (54%) had a high metastatic burden, and the metastatic burden was unknown for 122 (6%). Radiotherapy improved failure-free survival (HR 0.76, 95% CI 0.68-0.84) but not OS (HR 0.92, 95% CI 0.80-1.06). In a prespecified subgroup analysis, patients receiving radiotherapy to the prostate among patients with low metastatic burden, there was a significant improvement in OS (HR 0.68, 95% CI 0.52-0.90).
The SABR-COMET trial was an important trial published in 2019 assessing stereotactic body radiotherapy to oligometastatic disease.15 The objective of this study was to assess the standard of care of palliative treatments with or without stereotactic body radiotherapy in up to five metastatic lesions. The trialist’s hypothesis was that patients with oligometastatic disease will have improved outcomes with treatment of their metastatic sites. This study included any cancer (primarily breast, prostate, colorectal, and lung) who were then randomized 1:2 to standard of care versus standard of care plus stereotactic body radiotherapy. The standard of care was at the discretion of the physician and the primary endpoint was OS. There were 99 patients at 10 centers and median follow-up was 25.5 months. Median OS was 28 months (95% CI 19-33) in the control group versus 41 months (26-not reached) in the stereotactic body radiotherapy group (HR 0.57, 95% CI 0.30-1.10; p =0.090). Adverse events of grade 2 or worse occurred in three (9%) of 33 controls and 19 (29%) of 66 patients in the stereotactic body radiotherapy group (p=0.026), an absolute increase of 20% (95%CI 5-34). Treatment-related deaths occurred in three (4.5%) of 66 patients after stereotactic body radiotherapy, compared with none in the control group. The authors concluded that stereotactic body radiotherapy was associated with a 13-month increase in OS and doubling of progression-free survival (PFS).
Conclusions
The last five years have seen several important and practice-changing clinical trials for the treatment of prostate cancer with radiotherapy. Early results suggest that ultra-hypofractionation may make primary treatment with radiotherapy an attractive, less arduous option, however long-term OS results for hypofractionation will be important. There is little doubt that the addition of ADT to radiotherapy in high-risk patients for primary treatment and those experiencing biochemical failure after radical prostatectomy improves outcomes. Furthermore, based on results from the STAMPEDE Arm H clinical trial, many clinicians are now treating the prostate primary among men with low-volume metastatic disease. Finally, ongoing studies utilizing stereotactic body radiotherapy to metastatic sites will be important as we focus on improving quality as well as quantity of life for patients with advanced, heavily treated prostate cancer.
Published Date: January 2020
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Incrocci L, Wortel RC, Alemayehu WG, et al. Hypofractionated versus conventionally fractionated radiotherapy for patients with localised prostate cancer (HYPRO): final efficacy results from a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016;17(8):1061-1069.
2. Dearnaley D, Syndikus I, Mossop H, et al. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: 5-year outcomes of the randomised, non-inferiority, phase 3 CHHiP trial. Lancet Oncol. 2016;17(8):1047-1060.
3. Widmark A, Gunnlaugsson A, Beckman L, et al. Ultra-hypofractionated versus conventionally fractionated radiotherapy for prostate cancer: 5-year outcomes of the HYPO-RT-PC randomised, non-inferiority, phase 3 trial. Lancet. 2019;394(10196):385-395.
4. Michalski JM, Moughan J, Purdy J, et al. Effect of Standard vs Dose-Escalated Radiation Therapy for Patients With Intermediate-Risk Prostate Cancer: The NRG Oncology RTOG 0126 Randomized Clinical Trial. JAMA Oncol. 2018;4(6):e180039.
5. Zapatero A, Guerrero A, Maldonado X, et al. High-dose radiotherapy with short-term or long-term androgen deprivation in localised prostate cancer (DART01/05 GICOR): a randomised, controlled, phase 3 trial. Lancet Oncol. 2015;16(3):320-327.
6. Bolla M, Maingon P, Carrie C, et al. Short Androgen Suppression and Radiation Dose Escalation for Intermediate- and High-Risk Localized Prostate Cancer: Results of EORTC Trial 22991. J Clin Oncol. 2016;34(15):1748-1756.
7. D'Amico AV, Chen MH, Renshaw A, Loffredo M, Kantoff PW. Long-term Follow-up of a Randomized Trial of Radiation With or Without Androgen Deprivation Therapy for Localized Prostate Cancer. JAMA : the journal of the American Medical Association. 2015;314(12):1291-1293.
8. Denham JW, Joseph D, Lamb DS, et al. Short-term androgen suppression and radiotherapy versus intermediate-term androgen suppression and radiotherapy, with or without zoledronic acid, in men with locally advanced prostate cancer (TROG 03.04 RADAR): 10-year results from a randomised, phase 3, factorial trial. Lancet Oncol. 2019;20(2):267-281.
9. Nabid A, Carrier N, Martin AG, et al. Duration of Androgen Deprivation Therapy in High-risk Prostate Cancer: A Randomized Phase III Trial. Eur Urol. 2018;74(4):432-441.
10. Rosenthal SA, Hu C, Sartor O, et al. Effect of Chemotherapy With Docetaxel With Androgen Suppression and Radiotherapy for Localized High-Risk Prostate Cancer: The Randomized Phase III NRG Oncology RTOG 0521 Trial. J Clin Oncol. 2019;37(14):1159-1168.
11. Pollack A, Karrison TG, Balogh AG. Short term Androgen Deprivation Therapy Without or With Pelvic Lymph Node Treatment Added to Prostate Bed Only Salvage Radiotherapy: The NRG Oncology/RTOG 0534 SPPORT Trial. ASTRO. 2019.
12. Shipley WU, Seiferheld W, Lukka HR, et al. Radiation with or without Antiandrogen Therapy in Recurrent Prostate Cancer. N Engl J Med. 2017;376(5):417-428.
13. Boeve LMS, Hulshof M, Vis AN, et al. Effect on Survival of Androgen Deprivation Therapy Alone Compared to Androgen Deprivation Therapy Combined with Concurrent Radiation Therapy to the Prostate in Patients with Primary Bone Metastatic Prostate Cancer in a Prospective Randomised Clinical Trial: Data from the HORRAD Trial. Eur Urol. 2019;75(3):410-418.
14. Parker CC, James ND, Brawley CD, et al. Radiotherapy to the primary tumour for newly diagnosed, metastatic prostate cancer (STAMPEDE): a randomised controlled phase 3 trial. Lancet. 2018;392(10162):2353-2366.
15. Palma DA, Olson R, Harrow S, et al. Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (SABR-COMET): a randomised, phase 2, open-label trial. Lancet. 2019;393(10185):2051-2058.
Beyond First-line Treatment of Metastatic Castrate-resistant Prostate Cancer
Second-line treatment options for metastatic castrate-resistant prostate cancer
Cabazitaxel
Cabazitaxel is a new taxane drug with activity in docetaxel-resistant cancers. In the TROPIC study, a Phase III prospective randomized trial, cabazitaxel plus prednisone was compared to mitoxantrone plus prednisone in 755 mCRPC patients, who progressed after or during treatment with docetaxel2 (Figure 1). Patients received a maximum of ten cycles of cabazitaxel or mitoxantrone plus prednisone. Overall survival (OS) was the primary end-point, being significantly longer in cabazitaxel-treated patients (median: 15.1 vs. 12.7 months p < 0.0001). Progression-free survival (PFS) was significantly improved as well (median: 2.8 vs. 1.4 months, p < 0.0001), and prostate-specific antigen (PSA) response rate was also better (39.2% vs. 17.8%, p < 0.0002). Grade 3-4 adverse events developed more significantly in patients taking cabazitaxel, particularly hematological adverse effects (68.2% vs. 47.3%, p < 0.0002).3 Therefore, cabazitaxel should be given with prophylactic granulocyte colony-stimulating factor and needs to be administered by physicians with expertise in handling neutropenia and sepsis.4 When compared to docetaxel in the first-line setting, cabazitaxel was not shown to be superior.5
Figure 1. TROPIC study design
Abiraterone following docetaxel
The COU-AA-301 was a large Phase III randomized trial with a total of 1,195 mCRPC patients being randomised in a 2:1 ratio to abiraterone acetate plus prednisone or placebo plus prednisone (Figure 2). Abiraterone is an antiandrogen agent which inhibits the 17α-hydroxylase/C17,20-lyase (CYP17) enzyme. Initial positive results of this trial were reported after a median follow-up of 12.8 months6 and confirmed by the final analysis.7 All patients in this trial failed at least one chemotherapy regimen, which included docetaxel. The primary end-point was OS, and in the final analysis, after a median follow-up of 20.2 months there was a clear advantage to the abiraterone arm (median survival of 15.8 vs.11.2 months, HR: 0.74, p < 0.0001). The benefit for abiraterone remained in all secondary endpoints as well (PSA, radiologic tissue response, time to PSA or objective progression). No significant difference between the treatment arms was seen in the rate of grade 3-4 adverse events, aside from a higher rate of mineralocorticoid-related side-effects (mainly grade 1-2 fluid retention, edema, and hypokalaemia).7
Figure 2. COU-AA-301 study design
Enzalutamide after docetaxel
Enzalutamide after docetaxel
The AFFIRM trial randomized 1,199 mCRPC patients in a 2:1 fashion to enzalutamide, a nonsteroidal antiandrogen, or placebo (Figure 3). All accrued patients had progressed after docetaxel treatment.8 The planned interim analysis of the AFFIRM study was published in 2012 and after a median follow-up of 14.4 months, a clear benefit was shown for the enzalutamide-treated patients (median survival of 18.4 vs. 13.6 months, HR: 0.63, p < 0.001).8 This led to the recommendation to halt and unblind the study. Importantly, the observed benefit occurred irrespective of age, baseline pain intensity, and type of progression. Enzalutamide was also beneficial in patients with visceral metastases. The final analysis with longer follow-up had confirmed the OS results despite the crossover and extensive post-progression therapies. Enzalutamide also conferred a clear advantage in all the secondary endpoints (PSA, soft tissue response, quality of life, time to PSA or objective progression).8 No significant difference in the rate of side-effects was observed in the two groups, with a lower incidence of grade 3-4 adverse events in the enzalutamide arm. Importantly, enzalutamide-treated patients had a 0.6% incidence of seizures compared to none in the placebo arm.8
Figure 3. AFFIRM trial design
Apalutamide
Apalutamide
Radium-223
Radium-223 is a targeted alpha therapy and is the only bone-specific drug that has been associated with a survival benefit in the mCRPC space. The ALSYMPCA trial was a large Phase III trial accruing 921 symptomatic mCRPC patients, who failed or were unfit for docetaxel chemotherapy.13 In this trial, patients were randomized to six injections of radium-223 or placebo, plus standard of care in both arms (Figure 4). The primary end-point was OS, and radium-223 significantly improved median OS by 3.6 months (HR: 0.70, p < 0.001).13 Radium-223 also conferred prolonged time to first skeletal event, improvement in pain scores and quality of life.13 No significant difference was noted in the rate of adverse effects between the treatment arms, aside from slightly more haematologic toxicity and diarrhea with radium-223.13 Whether patients were pretreated with docetaxel did not affect the benefit and safety of radium-223.14 Due to safety concerns, the label of radium-223 was restricted to use after docetaxel and at least one AR targeted agent.15 Importantly, the ERA-223 study assessed the effectiveness of early use of radium-223 together with abiraterone acetate and prednisolone (Figure 5). Unfortunately, this trial showed significant safety risks, especially with fractures and more deaths. Therefore, this combination is currently not recommended. These safety risks were more significant in patients without the concurrent use of antiresorptive agents.16
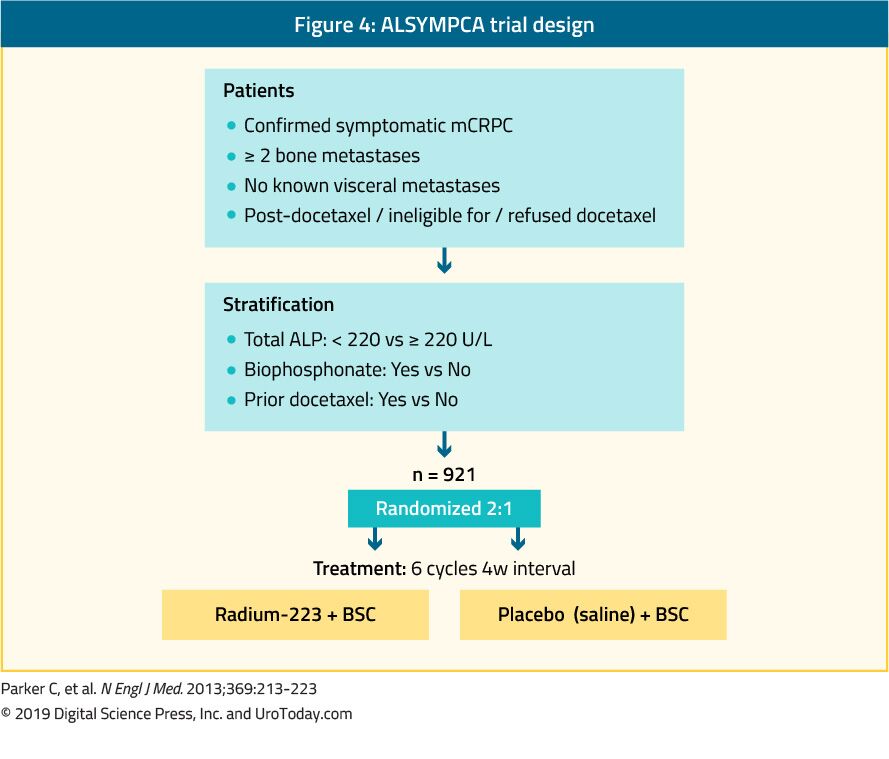
Figure 4. ALSYMPCA trial design
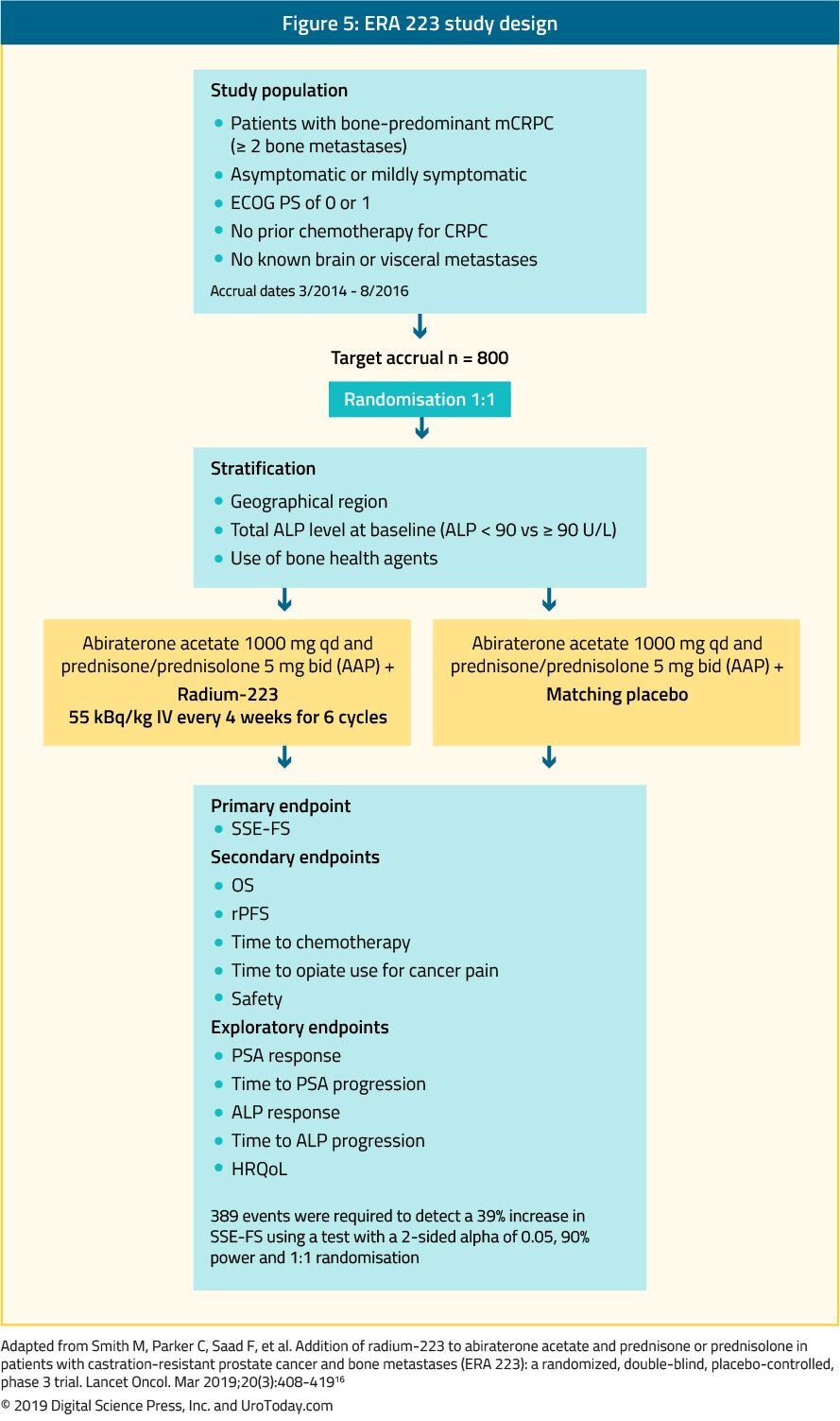
Figure 5. ERA 223 study design
Third line treatment following treatment with docetaxel and one hormonal treatment for metastatic castrate-resistant prostate cancer
Currently, there are no clear guidelines or recommendations regarding which treatment option is appropriate in this setting and this is open for debate. The choice for further treatment after docetaxel and one line of hormonal treatment for mCRPC is unclear.17 The available options include radium-223 or second-line chemotherapy (cabazitaxel). In unselected patients, subsequent treatments are expected to have a lower benefit than with earlier use18. There is also evidence that cross-resistance between enzalutamide and abiraterone exists.19, 20 There is a unique subset of patients worth mentioning with tumors demonstrating homozygous deletions or deleterious mutations in DNA-repair genes. In these patients Poly(ADP-ribose) polymerase (PARP) inhibitors have been reported to confer high rates of response. Therefore, patients who were previously treated with docetaxel and at least one novel hormonal agent; and whose tumors demonstrated homozygous deletions or deleterious mutations in DNA-repair genes showed an 88% response rate to Olaparib, a PAPR inhibitor.21 This represents an example of how treatment can be tailored according to the tumor mutation profile.22 In a randomized Phase II study of mCRPC patients, olaparib combined with abiraterone was compared to placebo and abiraterone. This study demonstrated a clinical benefit in olaparib-treated patients, regardless if mutations in DNA-repair genes existed.23 However, this combination treatment was shown to be toxic with significant side effects reported in 34% of patients vs. only 18% in the placebo arm.23
For patients with mismatch repair deficiency, the PD-1 inhibitor pembrolizumab was approved by the FDA for all tumors, including PCa. More specifically, pembrolizumab demonstrated antitumor activity and disease control with acceptable safety in RECIST-measurable and bone-predominant mCRPC, which was previously treated with docetaxel and novel AR antagonists.24
In the COMET-1 trial 1028 patients with progressive mCRPC after treatment with docetaxel and abiraterone and/or enzalutamide were randomly assigned at a 2:1 ratio to either cabozantinib 60 mg, a tyrosine kinase inhibitor, or prednisone 5 mg twice per day.25 The primary endpoint was OS, and the secondary endpoint included bone scan response after 12 weeks of treatment. Additional exploratory analyses included radiographic PFS (rPFS) and effects on circulating tumor cells, bone biomarkers, serum PSA, and symptomatic skeletal events.25 This trial demonstrated that cabozantinib did not significantly improve OS compared with prednisone in heavily pre-treated mCRPC patients (median OS was 11.0 months with cabozantinib and 9.8 months with prednisone, HR 0.90; 95% CI, 0.76 to 1.06; stratified log-rank P = 0.213).25 Cabozantinib had some activity in improving bone scan response, rPFS, symptomatic skeletal events, and bone biomarkers but not PSA outcomes.25
Changing and sequencing treatment in metastatic castrate-resistant prostate cancer
There are several open questions and dilemmas regarding when to change treatment in mCRPC patients and what is the most appropriate treatment sequence.
The appropriate time to change treatment in mCRPC patients is not entirely clear. No controversy exists regarding the need to change treatment when patients have symptomatic progression of their metastatic disease. Despite the many available treatment options to date, no head to head comparison has been made publicly available, while data assessing the correct sequence of treatment is being assessed. As data are lacking, physicians have been using the ECOG performance score to stratify patients before deciding on the “appropriate” treatment plan. Men with a good performance status are likely to tolerate more treatments as opposed to men with lower performance scores.
The National Comprehensive Cancer Network (NCCN) considers the onset of visceral disease to be a detrimental factor. Patients with liver metastases have especially poor outcomes for as of yet an unknown reason. In a meta-analysis including over 8,000 mCRPC patients who were enrolled in Phase III trials, patients with lymph-node- only disease appeared to have the best OS (median, 31.6 months; 95% CI, 27.9 to 36.6 months), with patients with lung and bone metastases having shorter and similar median OS (19.4 months [95% CI, 17.8 to 20.7 months] vs. 21.3 months [20.8 to 21.9], respectively), and patients with liver metastases demonstrating the worst OS (median, 13.5 months; 95% CI, 12.7 to 14.4 months).26 Therefore, the type of metastases the patient has can be used as a guide to when and how aggressive the treatment strategy should be.
Abiraterone and enzalutamide are highly active agents harboring a substantial effect on PFS, with trials comparing monotherapy with prednisone or placebo.27, 28 However, a subset of patients will not respond to these drugs. A patient who does not respond well will require a change of treatment. It is therefore important to see these patients frequently once starting therapy and assess their response. If no PSA decline is witnessed, the treatment needs to be changed.
When considering the appropriate treatment sequence in mCRPC, there are no clear guidelines or recommendations to date, and our limited knowledge is based mainly on retrospective data. In one non-randomized retrospective study, PFS, OS, and PSA responses from consecutive patients with chemotherapy-naïve mCRPC were compared between those who received abiraterone followed by enzalutamide and those who received enzalutamide followed by abiraterone.29 Initially, a slight improvement in patients who started with abiraterone and transitioned to enzalutamide was seen with improved PFS. An expanded retrospective study confirmed the general trend, showing that patients who started with abiraterone and then transitioned to enzalutamide had better PFS (median, 455 days [95% CI, 385 to 495 days]) than patients who started with enzalutamide and transitioned to abiraterone (median, 296 days; 95% CI, 235 to 358 days).30 However, OS was not significantly different between the groups.30 Furthermore, the authors of an ongoing randomized Phase II study comparing abiraterone vs. enzalutamide in patients with treatment-naïve mCRCP reported their interim results.31 After a median follow-up of 22.3 months, a PSA decline of more than 50% occurred in 34% of abiraterone treated patients compared to 4% in the enzalutamide treated patients (p<0.001).31 Additionally, the median time to PSA progression on 2nd-line therapy was 2.7 vs 1.3 months (HR 0.38, 95% CI 0.26-0.56) in favor of abiraterone.31 Lastly, the median OS was not reached vs 24.3 months (HR 0.82, 95% CI 0.53-1.27) in favor of abiraterone.31 As data regarding appropriate treatment sequencing is still being collected and analyzed, many physicians currently base their decision on which medication to start according to the adverse effects that we want to avoid. Abiraterone is commonly associated with edema, and therefore should be avoided in men with congestive heart failure,27 while enzalutamide is more likely to cause central nervous system toxicity and should probably be avoided in older patients.32
Radioligand therapy for metastatic castrate-resistant prostate cancer patients
PSMA-PET/CT imaging has significantly become more common in recent years. This has led to the emergence of a new field of radioligand directed therapy among heavily pretreated mCRPC patients. PCa metastases express PSMA, making it a promising approach to developing new tracers for targeted radionuclide therapies. PSMA is a non-secreted type II transmembrane protein produced almost exclusively by prostatic tissue and on tumor-associated neovasculature.33 Unlike other biomarkers, such as PSA, which may decrease with increasing neoplastic de-differentiation, PSMA has been shown to be upregulated in high-grade, de-differentiated PCa.34
Since 2015, several institutional studies have reported promising response rates and a favorable safety profile for radioligand therapy with 177Lu-PSMA-617 in mCRPC patients.35-37 However, these studies had small sample sizes and questionable generalizability. To addresses these limitations, a large multicenter German analysis assessed a cohort of patients treated with 177Lu-PSMA-617.38 This study included 145 mCRPC patients treated with 177Lu-PSMA-617 at 12 centers undergoing 1-4 therapy cycles. The study reported an overall biochemical response rate of 45% after all therapy cycles, with 40% of patients responding after a single cycle. Notably, negative predictors of the biochemical response included elevated alkaline phosphatase and the presence of visceral metastases.38
In a large meta-analysis published in 2017, 10 studies were assessed including 369 patients. This meta-analysis assessed the safety and efficacy of 177-Lutetium in mCRPC patients.39 The pooled proportion of patients with any PSA decline was 68% (95% CI: 61–74%); and the pooled proportion of patients with 450% PSA decline was 37% (95% CI: 22–52).39 This meta-analysis suggested promising early results for the treatment of mCRPC patients, especially in patients treated with the more recently developed radioligands, with approximately two-thirds of them showing a biochemical response.39
Although 177Lu-PSMA-617 is the most well-studied radioligand to date, there are additional compounds in development and undergoing initial testing. These include 177Lu-J591, 90Y-J591, 131I-MIP 1095, 177Lu-PSMA-I&T, and 225Ac-PSMA-617.40
Treatment and prevention of skeletal-related events
Patients with mCRPC commonly endure painful bone metastases with external beam radiotherapy (EBRT) being a highly effective treatment.41 Possible complications due to bone metastases include vertebral collapse or deformity, pathological fractures, and spinal cord compression. Cementation can be an effective treatment for a painful spinal fracture, clearly improving both pain and quality of life.42 However, standard palliative surgery can still be offered for managing osteoblastic metastases.43 Impending spinal cord compression is an emergency event that must be recognized as soon as possible. Patients should be educated to recognize the warning signs. If this is suspected, high-dose corticosteroids must be given and an MRI is required. A neurosurgeon or orthopedic surgeon consultation needs to be planned to discuss a possible decompression, followed by EBRT.44
Zoledronic acid, a bisphosphonate, has been evaluated in mCRPC patients in an attempt to reduce skeletal-related events (SRE). 643 mCRPC patients with bone metastases were randomized to receive zoledronic acid, 4 or 8 mg every three weeks for fifteen consecutive months, or placebo.45 The 8 mg dose was poorly tolerated without showing a significant benefit. However, at 15 and 24 months of follow-up, the 4 mg dose conferred fewer SREs compared to the placebo group (44 vs. 33%, p = 0.021), and less pathological fractures (13.1 vs. 22.1%, p = 0.015). Additionally, the time to first SRE was longer in the zoledronic acid group. However, no survival benefit was seen in any prospective trial assessing bisphosphonates.
Denosumab is a fully human monoclonal antibody directed against RANKL (receptor activator of nuclear factor kappa-B ligand). It is a key mediator of osteoclast formation, function, and survival. In non-metastatic CRPC, denosumab has been associated with increased bone-metastasis-free survival compared to placebo (median benefit: 4.2 months, HR: 0.85, p = 0.028).44 Like zoledronic acid, this benefit did not translate into a survival difference and neither the FDA or the EMA had approved denosumab for this indication.46 A Phase III trial compared the efficacy and safety of denosumab (n = 950) with zoledronic acid (n = 951) in mCRPC patients. Denosumab was shown to be superior to zoledronic acid in delaying or preventing SREs, as shown by time to first SRE (pathological fracture, radiation or surgery to bone, or spinal cord compression) of 20.7 vs. 17.1 months, respectively (HR: 0.82, p = 0.008). However, these findings were not associated with any survival benefit, and in a recent post-hoc re-evaluation of end-points, denosumab had actually shown an identical rate of SREs to zoledronic acid.47 It is critical to remember that these medications are associated with substantial toxicity, of 5% and 8.2% in non-metastatic CRPC and mCRPC, respectively.47, 48 All patients are required to be examined by a dentist prior to initiating this therapy, as the risk of jaw necrosis is increased by several risk factors including a history of trauma, dental surgery or dental infection.49 and the number of years the medication is used.
Recently, the randomized, double-blind Phase III trial (COMET-2; NCT01522443) was published, comparing cabozantinib, to mitoxantrone + prednisone in mCRPC patients with narcotic-dependent pain from bone metastases.50 All patients had progressed after treatment with docetaxel and either abiraterone or enzalutamide.50 The primary endpoint was pain response at week 6 and confirmed again at week 12. Enrollment was terminated early because cabozantinib did not demonstrate any survival benefit in mCRPC patients in the companion COMET-1 trial,25 described earlier. At study closure of the COMET-2 trial, only 119 patients were randomized. The trial demonstrated no significant difference in the pain response with cabozantinib versus mitoxantrone-prednisone.50
Future and ongoing trials
There are currently 24 registered ongoing Phase III trials involving mCRPC patients.
Some studies worth mentioning with much-anticipated results include the following:
- The combination of abiraterone and Olaparib as first-line therapy in mCRPC patients (NCT03732820)
- A study assessing the role of Rucaparib (a PARP inhibitor) vs. physician’s choice therapy in mCRPC patients (TRITON3 trial - NCT02975934)
- The combination of pembrolizumab with various other medications including enzalutamide (NCT03834493 - as part of the MK-3475-641/KEYNOTE-641 trial), docetaxel (NCT03834506 - as part of the MK-3475-921/KEYNOTE-921 trial), and olaparib (NCT03834519 – as part of the MK-7339-010/KEYLYNK-010)
- The ACIS trial, which will assess the combination of apalutamide, and abiraterone + prednisone in mCRPC patients (NCT02257736)
- A study assessing Masitinib (a tyrosine kinase inhibitor) plus docetaxel (NCT03761225)
- The combination of Talazoparib (a PARP inhibitor) + plus enzalutamide (NCT03395197),
- The combination of Atezolizumab (an anti-PD-L1 monoclonal antibody) + enzalutamide (NCT03016312)
- The combination of docetaxel and Radium-223 (NCT03574571)
- A study assessing 177Lu-PSMA-617 in mCRPC patients (NCT03511664)
- The IPATential150 trial – assessing the combination of Ipatasertib (an orally administered, ATP-competitive, selective AKT inhibitor) plus abiraterone (NCT03072238)
Conclusions
Substantial progress has been made in the mCRPC space in the last several years. Optimal management of mCRPC patients is a growing challenge as more potential treatments are added to the armamentarium. Choosing the right treatment for the right patient, and the correct sequence and combination of the increasing number of available medications will be the main challenge in the years to come. We currently lack level one evidence regarding the proper sequence and/or combination of current available medications, and physicians are faced with making these decisions without supporting data. Patients will most likely benefit from unique medications with complementary mechanisms of action in order to avoid cross-resistance. An important unmet clinical need thus far consists of acquiring evidence about the efficacy, safety, and tolerability of combination regimens, and optimized approaches for identifying patients most suited for specific treatments.
Published Date: November 19th, 2019
- Written by: Hanan Goldberg, MD
- References:
- Crawford ED, Petrylak DP, Shore N, et al. The Role of Therapeutic Layering in Optimizing Treatment for Patients With Castration-resistant Prostate Cancer (Prostate Cancer Radiographic Assessments for Detection of Advanced Recurrence II). Urology. Jun 2017;104:150-159.
- de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. Oct 2 2010;376(9747):1147-1154.
- Scher HI, Mazumdar M, Kelly WK. Clinical trials in relapsed prostate cancer: defining the target. J Natl Cancer Inst. Nov 20 1996;88(22):1623-1634.
- Resnick MJ, Lacchetti C, Penson DF. Prostate cancer survivorship care guidelines: American Society of Clinical Oncology practice guideline endorsement. J Oncol Pract. May 2015;11(3):e445-449.
- Oudard S, Fizazi K, Sengelov L, et al. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial-FIRSTANA. J Clin Oncol. Oct 1 2017;35(28):3189-3197.
- de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. New England Journal of Medicine. 2011;364(21):1995-2005.
- Fizazi K, Scher HI, Molina A, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. Oct 2012;13(10):983-992.
- Scher HI, Fizazi K, Saad F, et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. New England Journal of Medicine. 2012;367(13):1187-1197.
- Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J. Jan-Feb 2013;19(1):43-49.
- Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. Mar 15 2012;72(6):1494-1503.
- Rathkopf DE, Morris MJ, Fox JJ, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. Oct 1 2013;31(28):3525-3530.
- Rathkopf DE, Antonarakis ES, Shore ND, et al. Safety and Antitumor Activity of Apalutamide (ARN-509) in Metastatic Castration-Resistant Prostate Cancer with and without Prior Abiraterone Acetate and Prednisone. Clin Cancer Res. Jul 15 2017;23(14):3544-3551.
- Parker C, Nilsson S, Heinrich D, et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. New England Journal of Medicine. 2013;369(3):213-223.
- Hoskin P, Sartor O, O'Sullivan JM, et al. Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: a prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. Lancet Oncol. Nov 2014;15(12):1397-1406.
- European Medicines Agency (EMA). EMA restricts use of prostate cancer medicine Xofigo. https://www.ema.europa.eu/en/news/ema-restricts-use-prostate-cancer-medicine-xofigo.
- Smith M, Parker C, Saad F, et al. Addition of radium-223 to abiraterone acetate and prednisone or prednisolone in patients with castration-resistant prostate cancer and bone metastases (ERA 223): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. Mar 2019;20(3):408-419.
- de Bono JS, Smith MR, Saad F, et al. Subsequent Chemotherapy and Treatment Patterns After Abiraterone Acetate in Patients with Metastatic Castration-resistant Prostate Cancer: Post Hoc Analysis of COU-AA-302. Eur Urol. Apr 2017;71(4):656-664.
- Badrising S, van der Noort V, van Oort IM, et al. Clinical activity and tolerability of enzalutamide (MDV3100) in patients with metastatic, castration-resistant prostate cancer who progress after docetaxel and abiraterone treatment. Cancer. Apr 1 2014;120(7):968-975.
- Antonarakis ES, Lu C, Wang H, et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. New England Journal of Medicine. 2014;371(11):1028-1038.
- Attard G, Borre M, Gurney H, et al. Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer With Rising Prostate-Specific Antigen During Enzalutamide Treatment. J Clin Oncol. Sep 1 2018;36(25):2639-2646.
- Mateo J, Carreira S, Sandhu S, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. New England Journal of Medicine. 2015;373(18):1697-1708.
- Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine. 2015;372(26):2509-2520.
- Clarke N, Wiechno P, Alekseev B, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. Jul 2018;19(7):975-986.
- Antonarakis ES, Goh JC, Gross-Goupil M, et al. Pembrolizumab for metastatic castration-resistant prostate cancer (mCRPC) previously treated with docetaxel: Updated analysis of KEYNOTE-199. Journal of Clinical Oncology. 2019;37(7_suppl):216-216.
- Smith M, De Bono J, Sternberg C, et al. Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1. J Clin Oncol. Sep 1 2016;34(25):3005-3013.
- Halabi S, Kelly WK, Ma H, et al. Meta-Analysis Evaluating the Impact of Site of Metastasis on Overall Survival in Men With Castration-Resistant Prostate Cancer. J Clin Oncol. May 10 2016;34(14):1652-1659.
- Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. Jan 10 2013;368(2):138-148.
- Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. Jul 31 2014;371(5):424-433.
- Maughan BL, Luber B, Nadal R, Antonarakis ES. Comparing Sequencing of Abiraterone and Enzalutamide in Men With Metastatic Castration-Resistant Prostate Cancer: A Retrospective Study. Prostate. Jan 2017;77(1):33-40.
- Terada N, Maughan BL, Akamatsu S, et al. Exploring the optimal sequence of abiraterone and enzalutamide in patients with chemotherapy-naive castration-resistant prostate cancer: The Kyoto-Baltimore collaboration. Int J Urol. Jun 2017;24(6):441-448.
- Khalaf D, Annala M, Finch DL, et al. Phase 2 randomized cross-over trial of abiraterone + prednisone (ABI+P) vs enzalutamide (ENZ) for patients (pts) with metastatic castration resistant prostate cancer (mCPRC): Results for 2nd-line therapy. Journal of Clinical Oncology. 2018;36(15_suppl):5015-5015.
- Pilon D, Behl AS, Gozalo L, et al. Assessment of central nervous system (CNS) and dose reduction events in patients treated with abiraterone acetate plus prednisone (AA+P) or enzalutamide (ENZ). Journal of Clinical Oncology. 2016;34(15_suppl):5078-5078.
- Kulkarni HR, Singh A, Schuchardt C, et al. PSMA-Based Radioligand Therapy for Metastatic Castration-Resistant Prostate Cancer: The Bad Berka Experience Since 2013. J Nucl Med. Oct 2016;57(Suppl 3):97s-104s.
- Baum RP, Kulkarni HR, Schuchardt C, et al. 177Lu-Labeled Prostate-Specific Membrane Antigen Radioligand Therapy of Metastatic Castration-Resistant Prostate Cancer: Safety and Efficacy. J Nucl Med. Jul 2016;57(7):1006-1013.
- Ahmadzadehfar H, Eppard E, Kurpig S, et al. Therapeutic response and side effects of repeated radioligand therapy with 177Lu-PSMA-DKFZ-617 of castrate-resistant metastatic prostate cancer. Oncotarget. Mar 15 2016;7(11):12477-12488.
- Kratochwil C, Giesel FL, Stefanova M, et al. PSMA-Targeted Radionuclide Therapy of Metastatic Castration-Resistant Prostate Cancer with 177Lu-Labeled PSMA-617. J Nucl Med. Aug 2016;57(8):1170-1176.
- Rahbar K, Schmidt M, Heinzel A, et al. Response and Tolerability of a Single Dose of 177Lu-PSMA-617 in Patients with Metastatic Castration-Resistant Prostate Cancer: A Multicenter Retrospective Analysis. J Nucl Med. Sep 2016;57(9):1334-1338.
- Rahbar K, Ahmadzadehfar H, Kratochwil C, et al. German Multicenter Study Investigating 177Lu-PSMA-617 Radioligand Therapy in Advanced Prostate Cancer Patients. J Nucl Med. Jan 2017;58(1):85-90.
- Calopedos RJS, Chalasani V, Asher R, Emmett L, Woo HH. Lutetium-177-labelled anti-prostate-specific membrane antigen antibody and ligands for the treatment of metastatic castrate-resistant prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis. Sep 2017;20(3):352-360.
- Awang ZH, Essler M, Ahmadzadehfar H. Radioligand therapy of metastatic castration-resistant prostate cancer: current approaches. Radiat Oncol. May 23 2018;13(1):98.
- Dy SM, Asch SM, Naeim A, Sanati H, Walling A, Lorenz KA. Evidence-based standards for cancer pain management. J Clin Oncol. Aug 10 2008;26(23):3879-3885.
- Frankel BM, Monroe T, Wang C. Percutaneous vertebral augmentation: an elevation in adjacent-level fracture risk in kyphoplasty as compared with vertebroplasty. Spine J. Sep-Oct 2007;7(5):575-582.
- Frankel BM, Jones T, Wang C. Segmental polymethylmethacrylate-augmented pedicle screw fixation in patients with bone softening caused by osteoporosis and metastatic tumor involvement: a clinical evaluation. Neurosurgery. Sep 2007;61(3):531-537; discussion 537-538.
- Marco RA, Sheth DS, Boland PJ, Wunder JS, Siegel JA, Healey JH. Functional and oncological outcome of acetabular reconstruction for the treatment of metastatic disease. J Bone Joint Surg Am. May 2000;82(5):642-651.
- Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. Oct 2 2002;94(19):1458-1468.
- Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. Mar 5 2011;377(9768):813-822.
- Smith MR, Saad F, Coleman R, et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. Jan 7 2012;379(9810):39-46.
- Stopeck AT, Fizazi K, Body JJ, et al. Safety of long-term denosumab therapy: results from the open label extension phase of two phase 3 studies in patients with metastatic breast and prostate cancer. Support Care Cancer. Jan 2016;24(1):447-455.
- Aapro M, Abrahamsson PA, Body JJ, et al. Guidance on the use of bisphosphonates in solid tumours: recommendations of an international expert panel. Ann Oncol. Mar 2008;19(3):420-432.
- Basch EM, Scholz M, de Bono JS, et al. Cabozantinib Versus Mitoxantrone-prednisone in Symptomatic Metastatic Castration-resistant Prostate Cancer: A Randomized Phase 3 Trial with a Primary Pain Endpoint. Eur Urol. Jun 2019;75(6):929-937.
The Opioid Crisis in Urology
The Opioid Crisis in Urology
A study in 2011 from the University of Utah provided surveys to consecutive patients undergoing surgery during a 3-month time frame to assess perception of pain control, type and quantity of medication prescribed, quantity of leftover medication, refills needed, disposal instructions, and surplus medication disposition.2 Surveys were performed 2 to 4 weeks postoperatively, and with the exception of the investigators, prescribing physicians had no prior knowledge of the study. Among the 586 patients undergoing surgery, 47% participated in the study. Hydrocodone was prescribed most commonly (63%), followed by oxycodone (35%); 86% of the patients were satisfied with pain control. Of the dispensed narcotics, only 58% were consumed, while 12% of patients requested refills. A total of 67% of patients had surplus medication from the initial prescription and an alarming 92% received no disposal instructions for surplus medication. Among patients with leftover medication, 91% kept the medication at home while 6% threw it in the trash, 2% flushed it down the toilet, and less than 1% returned it to a pharmacy. Indeed, the retained surplus of medication provides a readily available source of opioid excess.In a prospective observational study of 155 opioid naïve patients who underwent a major prostate or kidney operation, investigators conducted a telephone survey 3-4 weeks postoperatively to assess the number of 5 mg oxycodone-equivalents prescribed, opioid use, and disposal.3 Most patients were male (86%), most were married (74%), the median was age 64 (IQR 59-70) years of age, and the majority were Caucasian (84%). Most patients reported social alcohol use (56%), but most denied current tobacco use (77%) or current and/or previous drug use (76%). Opioid prescribing exceeded use from 1.9- to 6.8-fold for all procedural categories. Overall, a total of 4,065 oxycodone-equivalents were prescribed during the study and 60% of pills prescribed went unused, resulting in 2,622 excess pills in the community.
Unfortunately, opioid overprescribing is not limited to the adult population, as it has also been demonstrated in pediatric urology patients. At the University of North Carolina, 117 pediatric urology patients’ parents were contacted with 39% completing a two-week post-operative telephone survey. The three most common pediatric urology procedures were inguinal hernia repair (n = 39), circumcision (n = 27), and cystoscopy (n = 16). Across all procedures, there was an average excess of 9.8 doses prescribed, corresponding to an over-prescription rate of 64%. Among the patients prescribed opioids, 41 (62%) had leftover opioid medication two weeks postoperatively. Thirty-two of 41 (78%) patients did not dispose of their leftover medication. Furthermore, only 13 patients received perioperative counseling on appropriate storage and disposal of opiates. A recent randomized control trial among 202 pediatric patients undergoing otolaryngologic or urologic procedures found that compared with providing only standard postoperative discharge instructions on opioid use, storage, and disposal, also providing a drug disposal bag significantly increased the rate of proper disposal of excess opioids by approximately 20%.4 These results suggest that a greater availability of disposal products may complement ongoing prescribing reduction efforts aimed at decreasing opioid misuse.
There are several reasons for the opioid crisis in urology, namely a culture of overprescribing.5 This may be due to:
- A historic failure to address acute pain in hospitalized patients, leading to the American Pain Society suggesting that pain should be akin to the fifth vital sign.6 Subsequently, physicians became more aware of their patients’ pain and were expected to treat their pain leading to an environment where liberal use of narcotics was tolerated.
- Over the past two decades, reimbursement, specifically through the Center for Medicare and Medicaid Services (CMS), has been linked to Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey. In the questionnaire, there were three questions dedicated to how well the patient’s pain was managed. These additional measures emphasizing the importance of pain management likely influenced the number of narcotics prescribed at discharge in order to maintain positive survey scores.
- Because narcotics must be prescribed via a hand-written prescription and obtaining additional pain medication is inconvenient for both patient and physician, providers may be more likely to overprescribe narcotics at discharge “just in case”.
The American Urological Association’s (AUA) Quality Improvement Summit on Opioid Stewardship in Urology
The AUA Quality Improvement Summit took place at AUA headquarters in December 2018 and was divided into four sessions:- Session 1: Physician-led Multicomponent Interventions in Opioid Stewardship. Dr. Richard Barth discussed procedure-specific opioid prescribing guidelines, Dr. Jonah Stulberg discussed opioid reclamation efforts, and Dr. Jim Dupree presented the Michigan MUSIC initiative on opioid stewardship.
- Session 2: Understanding Post-operative Pain. Dr. Brooke Chidgey discussed the pathophysiology of post-operative pain, Dr. Meghan Sperandeo-Fruge highlighted complementary alternative medicine pain management strategies, and Dr. Margaret Rukstalis discussed cognitive behavioral therapy and other non-pharmacologic approaches to pain management. In a sub-session discussing challenging cases in opioid management, Dr. Vernon Pais discussed the impact on prescription opioid use in patients with kidney stones, Dr. Matthew Nielsen highlighted the University of North Carolina Health Care System’s opioid stewardship program, and Dr. Benjamin Davies discussed his initiative of no opioids after a robotic prostatectomy.
- Session 3: High-Risk Patients and Expectations. Dr. Behfar Ehdaie presented on the expectation setting for opioid prescribing, Dr. Margaret Rukstalis discussed a surgeon’s role in the management of opioid misuse disorders, and Dr. Brooke Chidgey discussed the role of pain specialists for managing high-risk patients.
- Session 4: Policy and Outreach. Dr. Jennifer Waljee discussed opioid education and outreach, Dr. Scott Winiecki presented on opioid prescribing and the FDA safe use initiative, and Dr. Gregory Murphy completed the program discussing policy change and legislature to address the opioid crisis.
The full resources and slides for the AUA Quality Improvement Summit are available at: https://www.auanet.org/education/educational-calendar/quality-improvement-summit
Non-Opioid Measures for Pain Control
Data from Sweden suggests that opioid dependence may be specific to the U.S. Among 25,703 men in the National Prostate Cancer Register of Sweden who underwent radical prostatectomy, 16,368 men (64%) filled an opioid prescription during the 13 months before or after surgery.8 The use of strong opioids increased with time and the use of weak opioids decreased. There were 1.9% of men that had opioid prescriptions during the baseline period, followed by a spike to 59% around the time surgery, which sharply decreased by two months postoperatively. However, thereafter the proportion of men with opioid prescriptions remained slightly higher at 2.2% compared to the baseline before radical prostatectomy. Of chronic late users, 57% were previous users and 43% were new chronic users. Higher cancer risk category, greater comorbidity, unmarried status and low educational level were associated with the risk of new chronic opioid use. Although more than half of male Swedish patients filled an opioid prescription after radical prostatectomy, less than 1% of men became chronic opioid users.Professor Benjamin Davies from the University of Pittsburgh has been a thought leader and advocate for minimizing opioid prescriptions among patients undergoing urologic procedures, namely advocating for the “No Opioid Robotic Radical Prostatectomy”.9,10 This protocol is as follows:
- Pre-operative: Oral neurontin, acetaminophen, +/- celebrex
- Quadratus lumborum block (ropivicaine, decadron, precedex)
- Intraoperative: separate infusions of propofol, ketamine, and precede
- Post-operative: Toradol 15 mg IV PRN while in the hospital
- Tylenol and Motrin for 48 hours
Prospective Initiatives
In an effort to evaluate the effect of opioid reduction after radical prostatectomy on post-discharge opioid prescribing, use, and disposal, the ORIOLES trial was designed as a prospective, non-randomized, pre-post interventional trial.12 An evidence-based intervention included a discharge sheet, nursing education, and standardized prescribing guideline; the primary outcome was total oral morphine equivalents used after surgery. Secondary outcomes included opioid prescribing, opioid disposal, need for additional opioid medication, and presence of incisional/post-surgical abdominal pain beyond 30-days. There were 214 men in the pre-intervention arm and 229 men in the post-intervention arm with 100% follow-up. The intervention reduced post-discharge opioid prescribing from 224.3 mg to 120.3 mg (p=0.01), reduced opioid use from 52.1mg to 38.3mg (p<0.01), and increased opioid disposal by 13.5% (p<0.01). Greater post-discharge opioid use was associated with greater prescribing of opioids at discharge, higher body mass index, and use of opioid medication prior to surgery.From this prospective initiative, the authors demonstrate that a simple, three-component opioid reduction intervention was able to reduce opioid prescribing, reduce opioid use, and increase opioid disposal at 30-days after radical prostatectomy. Importantly, this prescribing guideline met the needs of 84% of patients, while only 2.2% of patients required additional opioid medication for pain. Furthermore, an impressive one-third of patients used no opioid pain medication after discharge.
Investigators from the Mount Sinai School of Medicine have also recently assessed the effect of implementing a nonopioid protocol for patients undergoing robotic-assisted radical cystectomy with extracorporeal urinary diversion.13 Among 52 patients undergoing surgery, patients received a multimodal pain management protocol, including a combination of nonopioid pain medications and regional anesthesia. These patients were compared to 41 patients undergoing robotic cystectomy prior to the implementation of the nonopioid protocol. In this study, the authors found that patients on the nonopioid protocol received a much lower dose of postoperative morphine milligram equivalents (2.5 vs. 44, p < 0.001), with no difference in pain scores. In the non-opioid protocol patients, the median time to regular diet was significantly shorter (4 days vs. 5 days, p = 0.002), and the length of stay was two days shorter compared to the control group (5 days vs. 7days, p < 0.001).
Conclusions
The urologic community has by no means been spared by the current opioid epidemic across the U.S. Several studies in both the adult and pediatric settings have demonstrated overprescribing measures with little to no counseling or options for appropriate disposal of opioids. Several measures are now in place to solve this problem5, including (i) greater utilization and implementation of Prescription Drug Monitoring Programs (PDMPs) to provide alerts to providers to patients who may be filling opiate prescriptions with multiple providers; (ii) CMS has removed the three questions from the HCAHPS survey related to pain control, effective January 2018; (iii) increased utilization of Enhanced Recovery After Surgery (ERAS) pathways as a measure for decreasing intra- and post-operative use of opioids; (iv) each of the 50 states have passed legislation to make readily available naloxone, which rapidly reverses the effects of opioids in the overdose setting; (v) the creation of procedure-specific guidelines for discharge opioid recommendations. For example, a Johns Hopkins expert panel assessing 20 common surgical procedures suggest that the ideal range of oxycodone 5-mg tablets prescribed to opioid naïve patients at discharge is 0-1014; (vi) the DEA sponsored “National Rx Takeback” initiative, providing collection sites (primarily pharmacies) for returning opioids. Certainly, the current opioid epidemic is multifactorial. However, judicious prescribing of opioids amongst the urology community is one actionable item that will make a difference for the betterment of our patients.Published Date: December 2019
- Written by: Zachary Klaassen, MD, MSc
- References:
References:
1. National Institute on Drug Abuse. Opioid Crisis. 2017. Available at: www.drugabuse.gov/drugs-abuse/opioids/opioid-crisis
2. Bates C, Laciak R, Southwick A, Bishoff J. Overprescription of postoperative narcotics: a look at postoperative pain medication delivery, consumption and disposal in urological practice. J Urol. 2011;185(2):551-555.
3. Theisen KM, Myrga JM, Hale N, et al. Excessive Opioid Prescribing After Major Urologic Procedures. Urology. 2019;123:101-107.
4. Lawrence AE, Carsel AJ, Leonhart KL, et al. Effect of Drug Disposal Bag Provision on Proper Disposal of Unused Opioids by Families of Pediatric Surgical Patients: A Randomized Clinical Trial. JAMA Pediatr. 2019:e191695.
5. Theisen K, Jacobs B, Macleod L, Davies B. The United States opioid epidemic: a review of the surgeon's contribution to it and health policy initiatives. BJU Int. 2018;122(5):754-759.
6. Quality improvement guidelines for the treatment of acute pain and cancer pain. American Pain Society Quality of Care Committee. JAMA. 1995;274(23):1874-1880.
7. Shah AS, Blackwell RH, Kuo PC, Gupta GN. Rates and Risk Factors for Opioid Dependence and Overdose after Urological Surgery. J Urol. 2017;198(5):1130-1136.
8. Loeb S, Cazzaniga W, Robinson D, Garmo H, Stattin P. Opioid Use After Radical Prostatectomy: Nationwide, Population Based Study in Sweden. J Urol. 2019:101097JU0000000000000451.
9. Theisen KM, Davies BJ. A Radical Proposition: Opioid-sparing Prostatectomy. Eur Urol Focus. 2019.
10. Pekala KR, Jacobs BL, Davies BJ. The Shrinking Grey Zone of Postoperative Narcotics in the Midst of the Opioid Crisis: The No-opioid Urologist. Eur Urol Focus. 2019.
11. Shahait M, Lee DI. Application of TAP Block in Laparoscopic Urological Surgery: Current Status and Future Directions. Curr Urol Rep. 2019;20(5):20.
12. Patel HD, Faisal FA, Patel ND, et al. Effect of a Prospective Opioid Reduction Intervention on Opioid Prescribing and Use after Radical Prostatectomy: Results of the ORIOLES Initiative. BJU Int. 2019.
13. Audenet F, Attalla K, Giordano M, et al. Prospective implementation of a nonopioid protocol for patients undergoing robot-assisted radical cystectomy with extracorporeal urinary diversion. Urol Oncol. 2019;37(5):300 e317-300 e323.
14. Overton HN, Hanna MN, Bruhn WE, et al. Opioid-Prescribing Guidelines for Common Surgical Procedures: An Expert Panel Consensus. J Am Coll Surg. 2018;227(4):411-418.
First-line Treatment for Metastatic Castrate-resistant Prostate Cancer
Regardless of the treatment given, approximately 20%-30% of patients with localized PCa progress to metastatic disease, commonly treated with hormonal therapy.5 This can be given through surgical castration (bilateral orchiectomy) or through medical castration using androgen deprivation therapy (ADT). Both methods achieve a castrate level of serum testosterone which is regarded as the standard of care for treating metastatic hormone-sensitive PCa (mHSPC). However, mHSPC is destined to progress to metastatic castrate-resistant prostate cancer (mCRPC).6 The castrate-resistant prostate cancer (CRPC) state is defined as disease progression despite reaching castrate testosterone levels (serum testosterone < 50 ng/dL or 1.7 nmol/L), and can present as either a continuous rise in serum PSA levels, progression of pre-existing disease, and/or the appearance of new metastases.7 CRPC has a median survival of approximately three years8 and is associated with a significant deterioration of quality of life.9 The exact mechanism of transition from mHSPC to mCRPC is still unclear. However, it is known that despite castrate levels of androgens, the androgen receptor (AR) remains active and continues to drive PCa progression in CRPC.10 This has led to the development of novel agents aimed at further decreasing androgen production or blocking AR function. However, there are other biologic pathways that function independently of androgen signaling and also result in CRPC.
Several significant shifts have occurred in the treatment options of the mHSPC space resulting in substantial survival benefit (please see “The rapidly evolving management strategy of metastatic Hormone-Sensitive Prostate Cancer” link), including the introduction of chemotherapy in the CHAARTED study11 and STAMPEDE trial,12 the addition of abiraterone acetate and prednisone in the LATITUDE study13 and STAMPEDE trial,14 the addition of enzalutamide in the ARCHES trial15 and the ENZAMET study,16 and lastly, the addition of apalutamide, an oral nonsteroidal anti-androgen, which like enzalutamide, binds directly to the ligand-binding domain of the AR and prevents AR translocation, DNA binding, and AR-mediated transcription.17 The TITAN trial showed overall survival (OS) benefit in apalutamide-treated mHSPC patients.18 Apalutamide has also shown benefit over placebo in the non-metastatic CRPC (nmCRP) setting in the SPARTAN phase 3 placebo-controlled trial,19 similar to the benefit shown by enzalutamide-treated nonmetastatic castrate-resistant prostate cancer (nmCRPC) patients, in the PROSPER trial20 (please see “The novel treatments for the non-metastatic castrate-resistant prostate cancer” link). These treatment advances in the mHSPC and nmCRPC setting have raised the question of which treatment options should be offered to patients in the mCRPC setting.21
The treatment of men with CRPC has dramatically changed over the last 15 years. Prior to 2004, when patients failed primary ADT, treatments were administered solely for palliation. The landmark trials by Tannock et al.22 and Petrylak et al.23 in 2004 were the first to introduce docetaxel chemotherapy in mCRPC patients that were shown to improve their survival. However, since docetaxel was FDA approved, five additional beneficial agents showing a survival advantage have been FDA-approved based on randomized clinical trials (Table 1). These include enzalutamide, and abiraterone, which specifically affect the androgen axis, sipuleucel-T, which stimulates the immune system;24 cabazitaxel, which is another chemotherapeutic agent;25 and radium-223, a radionuclide therapy.26 Other treatments for mCRPC have shown to improve outcomes but have yet been approved by the FDA and will be discussed in another review. Due to the substantial increase in multiple FDA-approved therapeutic agents in patients with CRPC, clinicians are challenged with a plethora of treatment options and multitude potential sequences of these agents, making clinical decision-making in mCRPC significantly more complex.
Table 1. Agents that have been approved for the treatment of metastatic castrate-resistant prostate cancer in the US
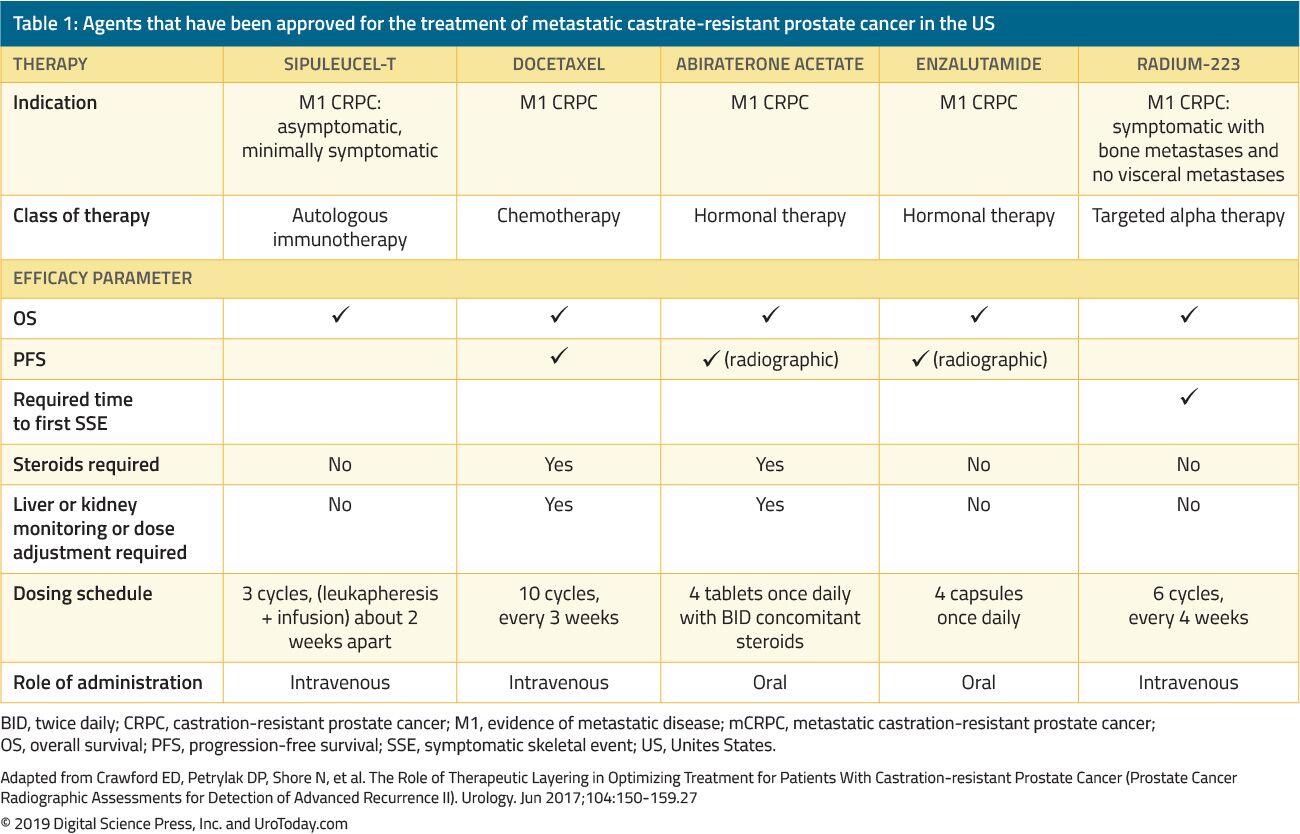
mCRPC is usually a debilitating disease, and patients will most likely benefit from a management strategy formalized by a multidisciplinary team consisting of urologists, medical oncologists, radiation oncologists, nurses, psychologists, and social workers.28 It is imperative to discuss palliation treatment options when considering additional systemic treatment, including management of pain, constipation, anorexia, nausea, depression, and fatigue.
Another crucial point to consider when establishing the appropriate treatment sequence in this disease space is the associated cost. Using models that included additional lines of treatment before or after docetaxel, the mean cost of mCRPC treatment during a mean period of 28.1 months was approximately $48,000 per patient.29 This cost is quite high due to the fact that patients may receive multiple lines of therapy and incur ongoing medical services during the course of their disease.30
Only two trials have demonstrated a marginal survival benefit for patients remaining on LHRH analogs instead of adding second- and third-line therapies.31, 32 Studies have shown that CRPC is not resistant to ADT, but rather hypersensitive to it.10 Treatment-mediated selection pressure during ADT causes the AR to amplify, and ensure the situation does not escalate, ADT is continued to be administered in the mCRPC setting. Treatment-mediated selection pressure also continues throughout the entire lifespan of the tumor, intensifying the need to correctly sequence therapies. However, because prospective data are lacking, the minute potential benefit of continuing castration still outweighs the minimal risk of this treatment. In addition, all subsequently approved treatments have been studied in men with ongoing ADT, adding another reason why it should be continued.
Before delving into the actual available treatment options, it is important to recognize that it is still unclear when to begin therapy in mCRPC patients who are completely asymptomatic. It is still unknown whether earlier treatment is superior, or if we should wait until the patient becomes symptomatic and develops pain. Before starting treatment, we should consider the patient’s existing comorbidities and expected adverse effects of starting therapy. Patients with early-stage mCRPC in the COU-AA-302 trial who received abiraterone typically survived almost one year longer than those who received placebo (median OS, 53.6 months vs. 41.8 months, respectively, HR, 0.61; 95% CI, 0.43 to 0.87; P = .006).33 Thus, early-stage mCRPC patients benefited from earlier start of abiraterone. In the same trial patients with asymptomatic or mildly symptomatic mCRPC, with baseline PSA < 15.6 ng/mL abiraterone also led to a faster rate and a greater degree of PSA decline than placebo.34 Although the currently available data is limited, it most likely suggests that starting treatment earlier rather than later is more advantageous.33, 34
Approved first-line treatment options for metastatic castrate-resistant prostate cancer
Abiraterone
Abiraterone is an antiandrogen which is an inhibitor of 17α-hydroxylase/C17,20-lyase (CYP17) enzyme. The COU-AA-302 phase III study evaluated abiraterone in 1,088 chemo-naïve, asymptomatic or mildly symptomatic mCRPC patients without visceral metastases. In this trial patients were randomized to abiraterone acetate or placebo, both combined with prednisone35 (Figure 1). Patients were stratified by either the Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1 and by asymptomatic or mildly symptomatic disease.35 OS and radiographic progression-free survival (rPFS) was the co-primary end-points. The trial demonstrated that after a median follow-up of 22.2 months, there was a significant improvement of rPFS in the abiraterone arm (median 16.5 vs. 8.2 months, HR 0.52, p < 0.001). At the final analysis after a median follow-up of 49.2 months, the OS end-point was significantly positive (34.7 vs. 30.3 months, HR: 0.81, 95% CI: 0.70-0.93, p = 0.0033).36 It is important to remember that mCRPC spans a broad prognostic spectrum even when it is chemotherapy-naïve.37 In an analysis of the abiraterone arm of the COU-AA-302 study, patients who had no pain at baseline, normal alkaline phosphatase and LDH levels, and less than 10 bone metastases had a median OS of 42.6 months.37 However, patients with more risk factors for progression had significantly shorter median OS.37 When assessing the toxicity profile of abiraterone, it seemed to confer more adverse events related to mineralocorticoid excess and liver function abnormalities, but these were mostly graded 1-2 adverse effects. Lastly, abiraterone was also shown to be equally effective in the elderly population (> 75 years).38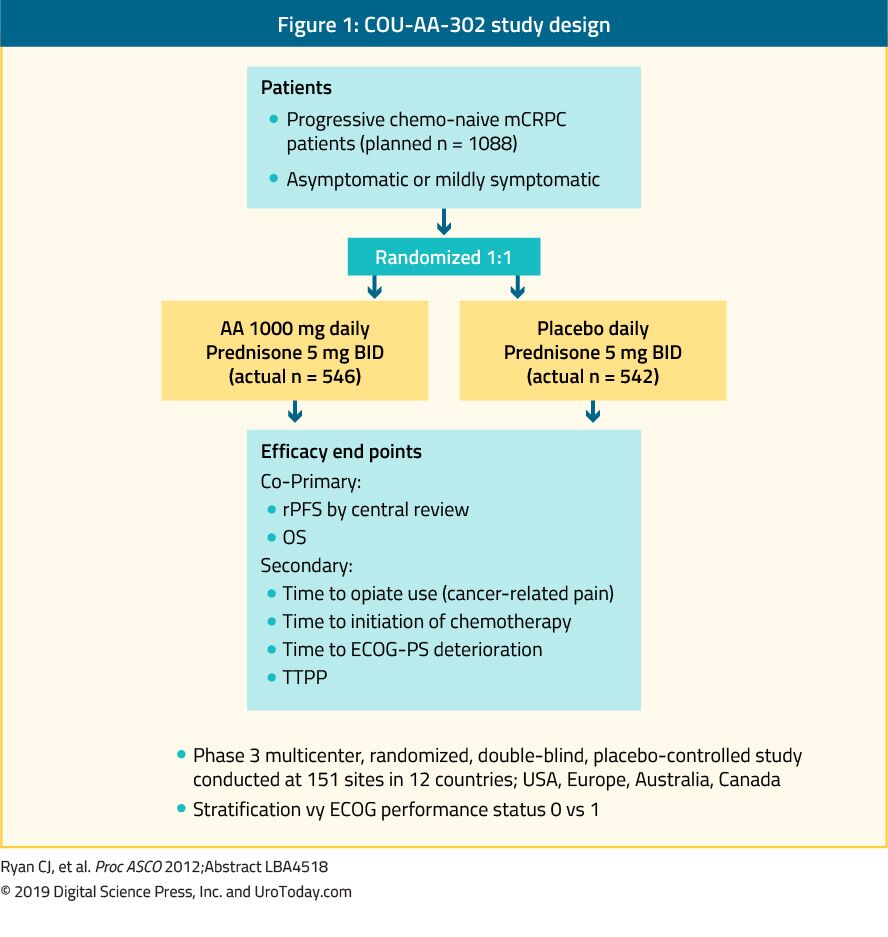
Figure 1. COU-AA-302 study design
Enzalutamide
Enzalutamide is a nonsteroidal antiandrogen. The PREVAIL study which is a randomized phase III trial included 1,717 chemo-naïve mCRPC patients and patients with visceral metastases were eligible as well.39 This trial compared enzalutamide to placebo (Figure 2). The PREVAIL trial showed a significant improvement in enzalutamide-treated patients in both co-primary endpoints, which included rPFS (HR: 0.186; CI: 0.15-0.23, p < 0.0001), and OS (HR: 0.706; CI: 0.6-0.84, p < 0.001). Extended follow-up and final analysis confirmed a benefit in OS and rPFS for enzalutamide.40 In 78% of patients treated with enzalutamide a PSA decrease of more than 50% was reported. The most common clinically relevant adverse events were fatigue and hypertension. Enzalutamide was also equally effective and well-tolerated in older men (> 75 years)41 and in those with or without visceral metastases.42 However, for men with liver metastases, there seemed to be no discernible benefit.43 The TERRAIN trial compared enzalutamide with bicalutamide, an older antiandrogen, in a randomized double-blind phase II study, showing a significant improvement in PFS (15.7 months vs. 5.8 months, HR: 0.44, p < 0.0001) in favor of enzalutamide.44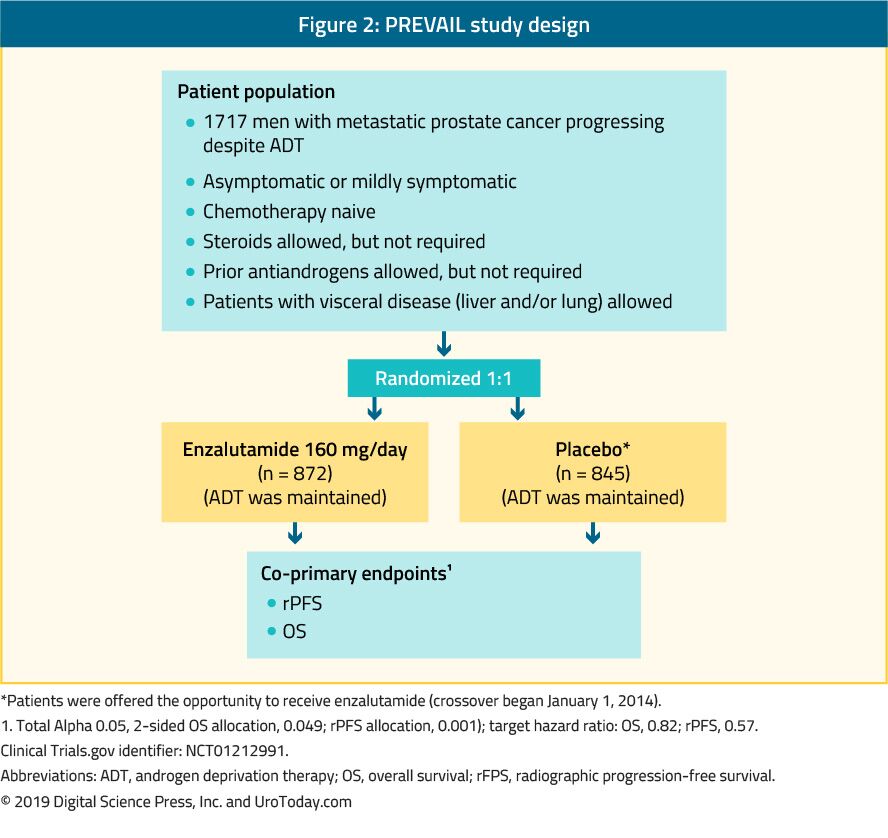
Figure 2. PREVAIL study design
Docetaxel
The landmark trial TAX 327 showed a significant improvement in median OS of 2-2.9 months in mCRPC patients treated with docetaxel-based chemotherapy when compared to patients who were treated with mitoxantrone plus prednisone therapy.22 The SWOG 9916 trial compared mitoxantrone to docetaxel and showed similar results23 (Figure 3). The standard first-line chemotherapy is docetaxel 75 mg/m2 in three-weekly doses combined with prednisone 5 mg twice a day, up to ten cycles. There are several important prognostic factors to consider when administering docetaxel: visceral metastases, pain, anemia (Hb < 13 g/dL), bone scan progression, and prior estramustine therapy. These prognostic factors may help to stratify response to docetaxel. Using these prognostic factors the disease can be categorized into low, intermediate and high risk, with significantly different corresponding median OS estimates of 25.7, 18.7 and 12.8 months, respectively.45 Although age by itself is not a contraindication to docetaxel therapy, patients must be fit enough to endure this type of treatment and comorbidities should be assessed prior to treatment initiation. In men who are thought to be unable to tolerate the standard dose and schedule of docetaxel, this can be decreased from 75 to 50 mg/m2 every two weeks, showing less grade 3-4 adverse events and a longer time to treatment failure.46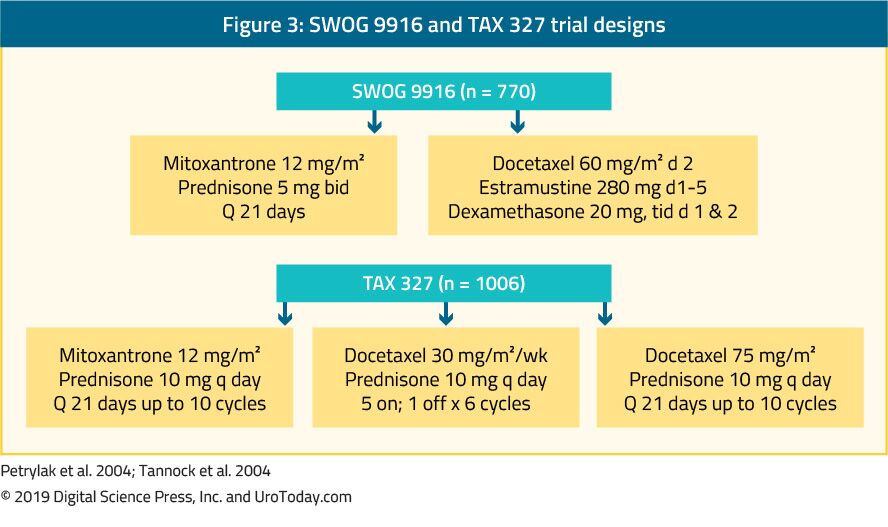
Figure 3. SWOG 9916 and TAX 327 trial designs
Sipuleucel-T
Sipuleucel-T, an autologous active cellular immunotherapy, was shown in a phase III trial (IMPACT trial) to confer a survival benefit in 512 asymptomatic or minimally symptomatic mCRPC patients when compared to placebo24 (Figure 4). After a median follow-up of 34 months, the median survival was significantly higher in the sipuleucel-T group (25.8 vs. 21.7 months, with an HR of 0.78,p = 0.03).24 Importantly, no PSA decline was observed during or after treatment and PFS was similar in both arms. The overall tolerance to sipuleucel-T was very good, with mostly grade 1-2 adverse events occurring. Currently, this treatment is only available in the US and is no longer available in Europe.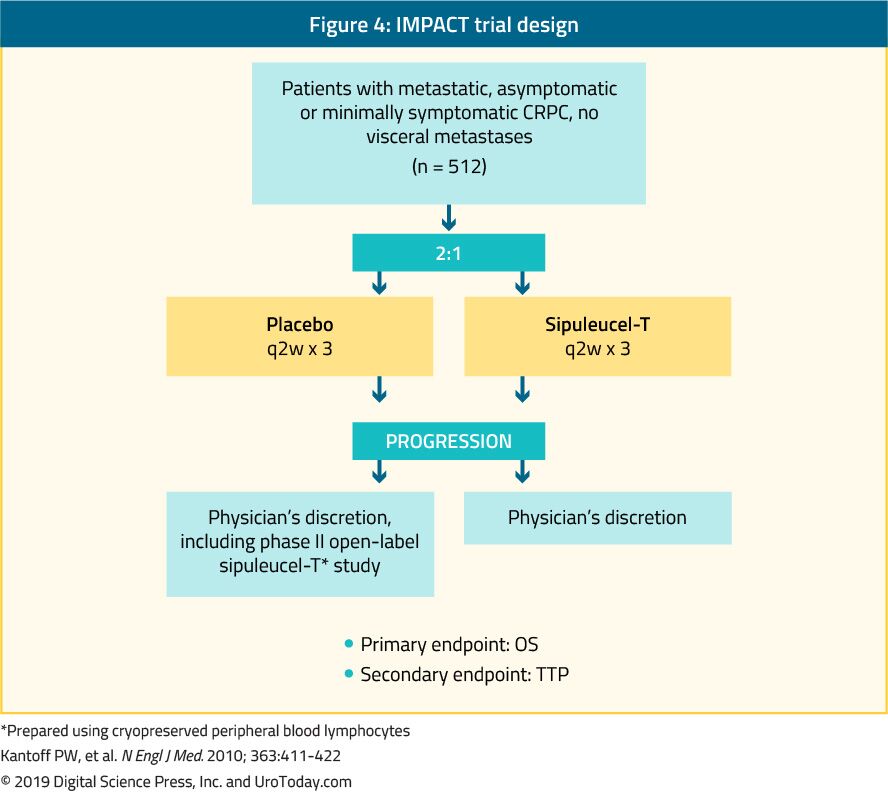
Figure 4. IMPACT trial design
Conclusions
In the last 15 years, there has been considerable scientific progress and investment in drug development for patients with mCRPC. This has resulted in the FDA approval of several lines of systemic therapies on grounds of pain palliation, minimizing disease adverse effects, and OS prolongation. To date, the reported impact on OS in mCRPC patients from each of these individual agents is still modest, resulting in an addition of only a few months. It is necessary to enhance our understanding of the disease biology of mCRPC, integrate a comprehensive molecular understanding of castration resistance, and analyze mechanisms of resistance to current therapies to improve future treatment development. It is also crucial to invest and develop predictive biomarkers to assist in the personalization of therapy. Lastly, on a more practical note, more data is needed on the appropriate second and third-line therapies, and sequencing and combination of available medications, discussed in more detail in the next review article (“Beyond first line treatment of metastatic castrate-resistant prostate cancer”).Published Date: November 19th, 2019
- Written by: Hanan Goldberg, MD
- References:
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: A Cancer Journal for Clinicians. 2019;69(1):7-34.
- Moyer VA. Screening for prostate cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. Jul 17 2012;157(2):120-134.
- Fedewa SA, Ward EM, Brawley O, Jemal A. Recent Patterns of Prostate-Specific Antigen Testing for Prostate Cancer Screening in the United States. JAMA Intern Med. Jul 1 2017;177(7):1040-1042.
- Negoita S, Feuer EJ, Mariotto A, Cronin KA, Petkov VI, Hussey SK. Annual Report to the Nation on the Status of Cancer, part II: Recent changes in prostate cancer trends and disease characteristics. Jul 1 2018;124(13):2801-2814.
- Amling CL, Blute ML, Bergstralh EJ, Seay TM, Slezak J, Zincke H. Long-term hazard of progression after radical prostatectomy for clinically localized prostate cancer: continued risk of biochemical failure after 5 years. J Urol. Jul 2000;164(1):101-105.
- Hotte SJ, Saad F. Current management of castrate-resistant prostate cancer. Curr Oncol. Sep 2010;17 Suppl 2:S72-79.
- Saad F, Hotte SJ. Guidelines for the management of castrate-resistant prostate cancer. Canadian Urological Association journal = Journal de l'Association des urologues du Canada. 2010;4(6):380-384.
- Tangen CM, Hussain MH, Higano CS, et al. Improved overall survival trends of men with newly diagnosed M1 prostate cancer: a SWOG phase III trial experience (S8494, S8894 and S9346). J Urol. Oct 2012;188(4):1164-1169.
- Damodaran S, Lang JM, Jarrard DF. Targeting Metastatic Hormone Sensitive Prostate Cancer: Chemohormonal Therapy and New Combinatorial Approaches. J Urol. May 2019;201(5):876-885.
- Mohler JL, Titus MA, Bai S, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. Feb 15 2011;71(4):1486-1496.
- Sweeney CJ, Chen Y-H, Carducci M, et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. New England Journal of Medicine. 2015;373(8):737-746.
- James ND, Sydes MR, Clarke NW, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. Mar 19 2016;387(10024):1163-1177.
- Fizazi K, Tran N, Fein L, et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. May 2019;20(5):686-700.
- James ND, de Bono JS, Spears MR, et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N Engl J Med. Jul 27 2017;377(4):338-351.
- Armstrong AJ, Szmulewitz RZ, Petrylak DP, et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J Clin Oncol. Jul 22 2019:Jco1900799.
- Davis ID, Martin AJ, Stockler MR, et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. New England Journal of Medicine. 2019;381(2):121-131.
- Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. Mar 15 2012;72(6):1494-1503.
- Chi KN, Agarwal N, Bjartell A, et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N Engl J Med. Jul 4 2019;381(1):13-24.
- Smith MR, Saad F, Chowdhury S, et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. New England Journal of Medicine. 2018;378(15):1408-1418.
- Hussain M, Fizazi K, Saad F, et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. New England Journal of Medicine. 2018;378(26):2465-2474.
- Gartrell BA, Saad F. Managing bone metastases and reducing skeletal related events in prostate cancer. Nat Rev Clin Oncol. Jun 2014;11(6):335-345.
- Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. New England Journal of Medicine. 2004;351(15):1502-1512.
- Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. Oct 7 2004;351(15):1513-1520.
- Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. Jul 29 2010;363(5):411-422.
- de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. Oct 2 2010;376(9747):1147-1154.
- Parker C, Nilsson S, Heinrich D, et al. Alpha Emitter Radium-223 and Survival in Metastatic Prostate Cancer. New England Journal of Medicine. 2013;369(3):213-223.
- Crawford ED, Petrylak DP, Shore N, et al. The Role of Therapeutic Layering in Optimizing Treatment for Patients With Castration-resistant Prostate Cancer (Prostate Cancer Radiographic Assessments for Detection of Advanced Recurrence II). Urology. Jun 2017;104:150-159.
- Esper PS, Pienta KJ. Supportive care in the patient with hormone refractory prostate cancer. Semin Urol Oncol. Feb 1997;15(1):56-64.
- Dragomir A, Dinea D, Vanhuyse M, Cury FL, Aprikian AG. Drug costs in the management of metastatic castration-resistant prostate cancer in Canada. BMC Health Serv Res. Jun 13 2014;14:252.
- Wen L, Valderrama A, Costantino ME, Simmons S. Real-World Treatment Patterns in Patients with Castrate-Resistant Prostate Cancer and Bone Metastases. Am Health Drug Benefits. May 2019;12(3):142-149.
- Hussain M, Wolf M, Marshall E, Crawford ED, Eisenberger M. Effects of continued androgen-deprivation therapy and other prognostic factors on response and survival in phase II chemotherapy trials for hormone-refractory prostate cancer: a Southwest Oncology Group report. J Clin Oncol. Sep 1994;12(9):1868-1875.
- Taylor CD, Elson P, Trump DL. Importance of continued testicular suppression in hormone-refractory prostate cancer. J Clin Oncol. Nov 1993;11(11):2167-2172.
- Miller K, Carles J, Gschwend JE, Van Poppel H, Diels J, Brookman-May SD. The Phase 3 COU-AA-302 Study of Abiraterone Acetate Plus Prednisone in Men with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer: Stratified Analysis Based on Pain, Prostate-specific Antigen, and Gleason Score. Eur Urol. Jul 2018;74(1):17-23.
- Ryan CJ, Londhe A, Molina A, et al. Relationship of baseline PSA and degree of PSA decline to radiographic progression-free survival (rPFS) in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (mCRPC): Results from COU-AA-302. Journal of Clinical Oncology. 2013;31(15_suppl):5010-5010.
- Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. Jan 10 2013;368(2):138-148.
- Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. Feb 2015;16(2):152-160.
- Ryan CJ, Kheoh T, Li J, et al. Prognostic Index Model for Progression-Free Survival in Chemotherapy-Naive Metastatic Castration-Resistant Prostate Cancer Treated With Abiraterone Acetate Plus Prednisone. Clin Genitourin Cancer. Jul 25 2017.
- Roviello G, Cappelletti MR, Zanotti L, et al. Targeting the androgenic pathway in elderly patients with castration-resistant prostate cancer: A meta-analysis of randomized trials. Medicine (Baltimore). Oct 2016;95(43):e4636.
- Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. Jul 31 2014;371(5):424-433.
- Beer TM, Armstrong AJ, Rathkopf D, et al. Enzalutamide in Men with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer: Extended Analysis of the Phase 3 PREVAIL Study. Eur Urol. Feb 2017;71(2):151-154.
- Graff JN, Baciarello G, Armstrong AJ, et al. Efficacy and safety of enzalutamide in patients 75 years or older with chemotherapy-naive metastatic castration-resistant prostate cancer: results from PREVAIL. Ann Oncol. Feb 2016;27(2):286-294.
- Evans CP, Higano CS, Keane T, et al. The PREVAIL Study: Primary Outcomes by Site and Extent of Baseline Disease for Enzalutamide-treated Men with Chemotherapy-naive Metastatic Castration-resistant Prostate Cancer. Eur Urol. Oct 2016;70(4):675-683.
- Alumkal JJ, Chowdhury S, Loriot Y, et al. Effect of Visceral Disease Site on Outcomes in Patients With Metastatic Castration-resistant Prostate Cancer Treated With Enzalutamide in the PREVAIL Trial. Clin Genitourin Cancer. Oct 2017;15(5):610-617.e613.
- Shore ND, Chowdhury S, Villers A, et al. Efficacy and safety of enzalutamide versus bicalutamide for patients with metastatic prostate cancer (TERRAIN): a randomised, double-blind, phase 2 study. Lancet Oncol. Feb 2016;17(2):153-163.
- Armstrong AJ, Garrett-Mayer E, de Wit R, Tannock I, Eisenberger M. Prediction of survival following first-line chemotherapy in men with castration-resistant metastatic prostate cancer. Clin Cancer Res. Jan 1 2010;16(1):203-211.
- Kellokumpu-Lehtinen PL, Harmenberg U, Joensuu T, et al. 2-Weekly versus 3-weekly docetaxel to treat castration-resistant advanced prostate cancer: a randomised, phase 3 trial. Lancet Oncol. Feb 2013;14(2):117-124.
The Genetics of Prostate Cancer
Germline mutations in prostate cancer carcinogenesis
Some of the first data to delineate the value of assessment of inherited genetic changes in prostate cancer came from Pritchard and colleagues who assessed the prevalence of mutations in 20 DNA-repair genes among 692 patients with metastatic prostate cancer8. They identified such mutations in 82 men (11.8%).
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Kang ZJ, Liu YF, Xu LZ, et al. The Philadelphia chromosome in leukemogenesis. Chin J Cancer 2016; 35:48.
2. An X, Tiwari AK, Sun Y, et al. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res 2010; 34(10):1255-68.
3. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343(2):78-85.
4. Stanford JL, Ostrander EA. Familial prostate cancer. Epidemiol Rev 2001; 23(1):19-23.
5. Carter BS, Bova GS, Beaty TH, et al. Hereditary prostate cancer: epidemiologic and clinical features. J Urol 1993; 150(3):797-802.
6. Bostwick DG, Burke HB, Djakiew D, et al. Human prostate cancer risk factors. Cancer 2004; 101(10 Suppl):2371-490.
7. Alvarez-Cubero MJ, Saiz M, Martinez-Gonzalez LJ, et al. Genetic analysis of the principal genes related to prostate cancer: a review. Urol Oncol 2013; 31(8):1419-29.
8. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 2016; 375(5):443-53.
9. Castro E, Romero-Laorden N, Del Pozo A, et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol 2019; 37(6):490-503.
10. Nicolosi P, Ledet E, Yang S, et al. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol 2019; 5(4):523-528.
11. Mateo J, Carreira S, Sandhu S, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 2015; 373(18):1697-708.
12. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015; 161(5):1215-1228.
13. Giri VN, Knudsen KE, Kelly WK, et al. Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J Clin Oncol 2018; 36(4):414-424.
14. Wallis CJ, Nam RK. Prostate Cancer Genetics: A Review. EJIFCC 2015; 26(2):79-91.
15. Ahmad AS, Vasiljevic N, Carter P, et al. A novel DNA methylation score accurately predicts death from prostate cancer in men with low to intermediate clinical risk factors. Oncotarget 2016; 7(44):71833-71840.
16. Majumdar S, Buckles E, Estrada J, et al. Aberrant DNA methylation and prostate cancer. Curr Genomics 2011; 12(7):486-505.
The Current Status of Immunotherapy for Prostate Cancer
The Prostate Cancer Immune Microenvironment
The microenvironment associated with prostate cancer includes low cytolytic activity of natural killer (NK) cells,5 high secretion of TGF-beta by prostate tissue (which inhibits NK and lymphocyte function),6 and recruitment of T regulatory cells that down-regulate antitumor immunity.7 As such, the prostate cancer microenvironment has been described as an immunosuppressive state. Furthermore, based on the chronicity of the prostate cancer disease spectrum, the immune microenvironment is likely dynamic, with changes over time/clinical states and with treatment exposure.8 For example, there are increased tumor-infiltrating lymphocytes in the prostate bed following androgen deprivation therapy,9 and higher levels of PD-1 ligand and PD-L2 expression on the surface of enzalutamide-treated prostate cancer cells.10 Several aspects make prostate cancer attractive for immunotherapy-based treatment options, including a high-level of tumor-associated antigens such as prostate-specific antigen (PSA), prostate acid phosphatase (PAP), and prostate-specific membrane antigen (PSMA).Cell-based vaccines
Cell-based vaccines consist of whole cells that are modified in order to induce anti-tumor immune responses. Sipuleucel-T is an autologous vaccine processed following peripheral dendritic cell collection via leukapheresis. This is then incubated with GCS-F and PAP protein, followed by reinfusion into the patient (after a 36-44 hour period) in order to generate a PAP-specific CD4+ and CD8+ T cell response.11,12Sipuleucel-T was FDA approved based on results of the Phase III IMPACT clinical trial.1 This trial enrolled 512 patients with mCRPC who had asymptomatic disease/minimally symptomatic with no visceral metastases, randomizing men to three infusions of sipuleucel-T (n=341) or placebo (n=171). The IMPACT trial noted a 4.1-month improvement in overall survival (OS) for those taking sipuleucel-T compared to placebo and a 22% reduction in risk of death. There was no difference between the groups with regards to objective disease progression or PSA response (secondary endpoints). An assessment of safety profile for patients in this study found that the treatment was overall well tolerated with minimal concern for severe adverse events.13 Furthermore, immunologic assessment showed that patients with high antibody titers against PA2024 benefited the most from treatment, noting longer survival.1 Despite the results and safety profile of IMPACT, reported nearly a decade ago, the use of sipuleucel-T has not been widely adopted primarily due to the lack of cost-effectiveness and the infrastructure required to administer this treatment.
Vector-based vaccines
Vector-based vaccines consist of genetically engineered nucleic acids that encode specific tumor-associated antigens transmitted by vectors such as bacterial plasmids or viruses. DNA-based vaccines can be incorporated by host cells and generate an immune response to recruiting antigen-presenting cells. pTVG-HP is a DNA plasmid vector vaccine that encodes PAP protein. pTVG-HP has been tested in the non-metastatic CRPC setting, demonstrating increased PSA doubling time from 6.5 months to 9.3 months after one year of treatment.14PROSTVAC is a PSA-target pox-virus-based vaccine. PROSTVAC was tested in a Phase II study of 125 patients with minimally symptomatic mCRPC who were randomized to receive the vaccine or placebo.15 Although the study was negative for its primary endpoint of progression-free survival (PFS), OS after 3 years of follow-up was significantly increased by 8.5 months (25.1 vs 16.6 months; HR 0.56; p=0.0061).
PROSTVAC was subsequently tested in a Phase III trial that reported results earlier this year.16 Patients were randomly assigned to PROSTVAC (n = 432), PROSTVAC plus granulocyte-macrophage colony-stimulating (GMCS) factor (n = 432), or placebo (n = 433), stratified by PSA (< 50 ng/mL vs. >= 50 ng/mL) and lactate dehydrogenase (< 200 vs >= 200 U/L). The primary endpoint for this trial was OS, and secondary endpoints were patients alive without events (AWE): radiographic progression, pain progression, chemotherapy initiation, or death at 6 months. Unfortunately, neither active treatment had an effect on median OS: (i) PROSTVAC: 34.4 months, hazard ratio (HR) 1.01, 95% confidence interval (CI) 0.84-1.20 (ii) PROSTVAC plus GMCS factor: 33.2 months, HR 1.02, 95% CI 0.86-1.22 (iii) placebo: 34.3 months. Furthermore, AWE at 6 months was similar between the arms. Based on these results, the authors noted that focus is currently ongoing for combination therapies.
DCVAC/PCa is an autologous dendritic cell vaccine derived from mononuclear cells that are pulsed with killed prostate cancer cells. In a Phase I/II trial, there were 25 men with mCRPC that received DCVAC/PCa plus docetaxel, demonstrating good tolerability and a median OS of 19 months.17 Currently, there is a Phase III (VIABLE) trial of DCVAC/PCa ongoing, which began accrual in 2014, with a target of 1,170 patients and planned completion date in 2020.
Immune Checkpoint inhibitors
Checkpoint inhibitors are antibodies that target molecules, such as cytotoxic T-lymphocyte protein 4 (CTLA-4) or PD-1 and its ligand PD-L1. Among men with mCRPC, ipilimumab was tested in the Phase III for those who had progressed on docetaxel chemotherapy, randomizing 799 patients to ipilimumab or placebo after bone-directed radiotherapy.18 The primary endpoint was OS, with no difference between the groups (ipilimumab 11.2 months vs placebo 10 months; HR 0.85, p=0.053); however, there was a small benefit in PFS favoring ipilimumab (4.0 vs 3.1 months; HR 0.70, p < 0.0001). More recently, Beer et al.19 reported findings of another Phase III trial randomizing 602 patients (2:1) with metastatic chemotherapy-naïve CRPC to ipilimumab vs placebo. Similar to the post-docetaxel patients, there was no difference in OS between the groups (HR 1.11, 95% CI 0.88-1.39), however men receiving ipilimumab had improved PFS (5.6 months vs 3.8; HR 0.67, 95% CI 0.55-0.81) compared to those receiving placebo.Pembrolizumab has recently moved into the mCRPC arena, receiving FDA approval in a tumor agnostic indication for MSI-high (MSI-H) mutation CRPC patients in 2017. A study from the Memorial Sloan Kettering Cancer Center assessed the prevalence of MSI-H/dMMR prostate cancer among 1,033 patients treated at their institution,20 finding that 32 (3.1%) had MSI-H/dMMR disease. This included 23 patients (2.2%) that had tumors with high MSIsensor scores, and 7 of the 32 MSI-H/dMMR patients (21.9%) with pathogenic germline mutation in a Lynch syndrome-associated gene. Eleven patients with MSI-H/dMMR CRPC received anti-PD-1/PD-L1 therapy and six of these had a greater than 50% decline in PSA levels. Based on these data, experts in the field of advanced prostate cancer feel that every mCRPC patient should be tested for MSI-H status and potential pembrolizumab eligibility.
The KEYNOTE-028 study was a trial of pembrolizumab in advanced solid tumors among patients with PD-1 expression ≥1% of tumor or stromal cells. Among 245 men screened, there were 35 PD-1% (14.3%) and 23 patients who enrolled.21 There were four partial responses, for an objective response rate of 17.4% and 8 of 23 (34.8%) patients had stable disease. Median duration of response was 13.5 months, and median PFS and OS were 3.5 and 7.9 months, respectively. Furthermore, the 6-month PFS and OS rates were 34.8% and 73.4%, respectively. Recently, off-label use of pembrolizumab among a heavily pre-treated population of mCRPC patients has recently been reported. At the 2019 ASCO GU meeting, Tucker and colleagues presented data on 51 patients, 86% of which had received three or more prior lines of therapy. Most patients had previously received abiraterone (88%), docetaxel (86%), enzalutamide (80%), and sipuleucel-T (74%). Among these patients, 16% had a >50% confirmed PSA decline with pembrolizumab, with 8% having >90% PSA decline. Fifty-nine percent of men were treated with some form of concurrent therapy along with pembrolizumab, most commonly enzalutamide (47%).
At the 2019 ASCO GU meeting, results of the Phase II KEYNOTE-650 were also presented. This trial tested the combination of nivolumab plus ipilimumab for men with mCRPC. There were two cohorts for this study: cohort 1 – asymptomatic or minimally symptomatic, who had progressed after at least 1 second generation hormone therapy with no prior chemotherapy, and cohort 2 – progression after chemotherapy. Overall response rates were 26% in cohort 1 and 10% in cohort 2, including two patients in each cohort who had a complete response. Median time to response was approximately two months. PSA response rate was 18% in cohort 1 and 10% in cohort 2.
Future Directions
Unlike many other tumor sites, to date, there has not been robust data to demonstrate a large role for immunotherapy in patients with mCRPC. However, there are several potential ways to increase immunotherapy response with the goal of improving the outcomes of immunotherapy for prostate cancer: (i) combination therapy, (ii) immune modulation, (iii) biomarkers for improving patient selection.Options for combination therapies:
1) Combination of immunotherapies: multiple vaccines, vaccine plus an immune checkpoint inhibitor, or an immunocytokine plus an immune checkpoint inhibitor. KEYNOTE-650 combining nivolumab plus ipilimumab is an example of improved efficacy among patients receiving combination therapy.
2) Combinations with therapies to capitalize on immunologic synergy: these studies assess the effect of the addition of other accepted treatments such enzalutamide, poly ADP ribose polymerase PARP inhibitors, radium-223, and docetaxel to immunotherapy regimens.
3) Given the changing microenvironment of prostate cancer across disease states, beginning combination immunotherapy earlier (castration-sensitive) may improve the immunotherapeutic benefit.
There are several options of immunomodulatory agents that target the tumor microenvironment to improve immunotherapy efficacy. Docetaxel has been proven to induce immunogenic modulations, such as increasing expression of ICAM-1, MUC-1, and MHC class 1 molecules.22 Additionally, tasquinimod is an immunomodulatory agent that blocks S100A9, a key regulatory molecule of myeloid cells. In a Phase II trial of 206 asymptomatic chemotherapy-naïve mCRPC patients randomized to tasquinimod vs placebo, men receiving tasquinimod had significantly improved disease progression (7.6 vs 3.3 months, p = 0.0042).23 Unfortunately, a Phase III trial assessing tasquinimod did not improve OS (21.3 for tasquinimod vs 24 months for placebo, HR 1.10, p=0.25), however, there was an improvement in radiographic PFS (7.0 months vs 4.4 months, HR 0.64, p = 0.001).24
Biomarkers continue to be an active area of research, not just for the selection of appropriate patients for immunotherapy, but also for other treatment regimens for advanced prostate cancer (ie. BRCA status for selecting patients for PARP inhibitors).
As follows is a summary of several of the current biomarkers as related to immunotherapy and prostate cancer (adapted from Maia and Hansen8):

As follows is a summary of ongoing, recruiting phase III trial assessing immunotherapy in prostate cancer:
Conclusion
To date, immunotherapy in prostate cancer has been less successful than other cancer types, with only Sipuleucel-T demonstrating an OS advantage of 4.1 months in a Phase III trial. Given the plethora of other treatment options, Sipuleucel-T is uncommonly used. However, with improved combination therapy, immunomodulation and biomarkers in addition to ongoing Phase III trials, there are additional assessments and results in upcoming that may improve the immunotherapy landscape and add to the armamentarium of treatment options for men with advanced prostate cancer.
Published Date: November 2019
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411-422.
2. Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16(2):152-160.
3. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147-1154.
4. Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213-223.
5. Pasero C, Gravis G, Guerin M, et al. Inherent and Tumor-Driven Immune Tolerance in the Prostate Microenvironment Impairs Natural Killer Cell Antitumor Activity. Cancer Res. 2016;76(8):2153-2165.
6. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554-567.
7. Sfanos KS, Bruno TC, Maris CH, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res. 2008;14(11):3254-3261.
8. Maia MC, Hansen AR. A comprehensive review of immunotherapies in prostate cancer. Crit Rev Oncol Hematol. 2017;113:292-303.
9. Thoma C. Prostate cancer: Towards effective combination of ADT and immunotherapy. Nat Rev Urol. 2016;13(6):300.
10. Bishop JL, Sio A, Angeles A, et al. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget. 2015;6(1):234-242.
11. Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10(8):580-593.
12. Ren R, Koti M, Hamilton T, et al. A primer on tumour immunology and prostate cancer immunotherapy. Can Urol Assoc J. 2016;10(1-2):60-65.
13. Hall SJ, Klotz L, Pantuck AJ, et al. Integrated safety data from 4 randomized, double-blind, controlled trials of autologous cellular immunotherapy with sipuleucel-T in patients with prostate cancer. J Urol. 2011;186(3):877-881.
14. McNeel DG, Dunphy EJ, Davies JG, et al. Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J Clin Oncol. 2009;27(25):4047-4054.
15. Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099-1105.
16. Gulley JL, Borre M, Vogelzang NJ, et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2019;37(13):1051-1061.
17. Podrazil M, Horvath R, Becht E, et al. Phase I/II clinical trial of dendritic-cell based immunotherapy (DCVAC/PCa) combined with chemotherapy in patients with metastatic, castration-resistant prostate cancer. Oncotarget. 2015;6(20):18192-18205.
18. Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700-712.
19. Beer TM, Kwon ED, Drake CG, et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J Clin Oncol. 2017;35(1):40-47.
20. Abida W, Cheng ML, Armenia J, et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2018.
21. Hansen AR, Massard C, Ott PA, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann Oncol. 2018;29(8):1807-1813.
22. Hodge JW, Garnett CT, Farsaci B, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. 2013;133(3):624-636.
23. Pili R, Haggman M, Stadler WM, et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol. 2011;29(30):4022-4028.
24. Sternberg C, Armstrong A, Pili R, et al. Randomized, Double-Blind, Placebo-Controlled Phase III Study of Tasquinimod in Men With Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2016;34(22):2636-2643.
Oligometastatic Prostate Cancer – Treatment of the Primary Tumor and Metastasis Directed Therapy
- Written by: Hanan Goldberg, MD
- References:
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 2018; 68(6): 394-424.
2. Hellman S, Weichselbaum RR. Oligometastases. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1995; 13(1): 8-10.
3. Weichselbaum RR, Hellman S. Oligometastases revisited. Nature reviews Clinical oncology 2011; 8(6): 378-82.
4. Soloway MS, Hardeman SW, Hickey D, et al. Stratification of patients with metastatic prostate cancer based on extent of disease on initial bone scan. Cancer 1988; 61(1): 195-202.
5. Weiner AB, Nettey OS, Morgans AK. Management of Metastatic Hormone-Sensitive Prostate Cancer (mHSPC): an Evolving Treatment Paradigm. Current treatment options in oncology 2019; 20(9): 69.
6. Rao A, Vapiwala N, Schaeffer EM, Ryan CJ. Oligometastatic Prostate Cancer: A Shrinking Subset or an Opportunity for Cure? American Society of Clinical Oncology educational book American Society of Clinical Oncology Annual Meeting 2019; 39: 309-20.
7. Tosoian JJ, Gorin MA, Ross AE, Pienta KJ, Tran PT, Schaeffer EM. Oligometastatic prostate cancer: definitions, clinical outcomes, and treatment considerations. Nature reviews Urology 2017; 14(1): 15-25.
8. Foster CC, Weichselbaum RR, Pitroda SP. Oligometastatic prostate cancer: Reality or figment of imagination? Cancer 2019; 125(3): 340-52.
9. Triggiani L, Alongi F, Buglione M, et al. Efficacy of stereotactic body radiotherapy in oligorecurrent and in oligoprogressive prostate cancer: new evidence from a multicentric study. British journal of cancer 2017; 116(12): 1520-5.
10. Pembroke CA, Fortin B, Kopek N. Comparison of survival and prognostic factors in patients treated with stereotactic body radiotherapy for oligometastases or oligoprogression. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology 2018; 127(3): 493-500.
11. Jorgensen T, Muller C, Kaalhus O, Danielsen HE, Tveter KJ. Extent of disease based on initial bone scan: important prognostic predictor for patients with metastatic prostatic cancer. Experience from the Scandinavian Prostatic Cancer Group Study No. 2 (SPCG-2). European urology 1995; 28(1): 40-6.
12. Gandaglia G, Karakiewicz PI, Briganti A, et al. Impact of the Site of Metastases on Survival in Patients with Metastatic Prostate Cancer. European urology 2015; 68(2): 325-34.
13. Gakis G, Boorjian SA, Briganti A, et al. The role of radical prostatectomy and lymph node dissection in lymph node-positive prostate cancer: a systematic review of the literature. European urology 2014; 66(2): 191-9.
14. von Bodman C, Godoy G, Chade DC, et al. Predicting biochemical recurrence-free survival for patients with positive pelvic lymph nodes at radical prostatectomy. The Journal of urology 2010; 184(1): 143-8.
15. Briganti A, Karnes JR, Da Pozzo LF, et al. Two positive nodes represent a significant cut-off value for cancer specific survival in patients with node positive prostate cancer. A new proposal based on a two-institution experience on 703 consecutive N+ patients treated with radical prostatectomy, extended pelvic lymph node dissection and adjuvant therapy. European urology 2009; 55(2): 261-70.
16. Francini E, Gray KP, Xie W, et al. Time of metastatic disease presentation and volume of disease are prognostic for metastatic hormone sensitive prostate cancer (mHSPC). 2018; 78(12): 889-95.
17. Linch M, Goh G, Hiley C, et al. Intratumoural evolutionary landscape of high-risk prostate cancer: the PROGENY study of genomic and immune parameters. Annals of oncology : official journal of the European Society for Medical Oncology 2017; 28(10): 2472-80.
18. Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015; 520(7547): 353-7.
19. Cooper CS, Eeles R, Wedge DC, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nature genetics 2015; 47(4): 367-72.
20. Larbi A, Dallaudiere B, Pasoglou V, et al. Whole body MRI (WB-MRI) assessment of metastatic spread in prostate cancer: Therapeutic perspectives on targeted management of oligometastatic disease. The Prostate 2016; 76(11): 1024-33.
21. Graziani T, Ceci F, Castellucci P, et al. (11)C-Choline PET/CT for restaging prostate cancer. Results from 4,426 scans in a single-centre patient series. European journal of nuclear medicine and molecular imaging 2016; 43(11): 1971-9.
22. McAllister SS, Gifford AM, Greiner AL, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 2008; 133(6): 994-1005.
23. Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005; 438(7069): 820-7.
24. Bayne CE, Williams SB, Cooperberg MR, et al. Treatment of the Primary Tumor in Metastatic Prostate Cancer: Current Concepts and Future Perspectives. European urology 2016; 69(5): 775-87.
25. Locke JA, Dal Pra A, Supiot S, Warde P, Bristow RG. Synergistic action of image-guided radiotherapy and androgen deprivation therapy. Nature reviews Urology 2015; 12(4): 193-204.
26. Kalina JL, Neilson DS, Comber AP, et al. Immune Modulation by Androgen Deprivation and Radiation Therapy: Implications for Prostate Cancer Immunotherapy. Cancers (Basel) 2017; 9(2): 13.
27. Heidenreich A, Pfister D, Porres D. Cytoreductive radical prostatectomy in patients with prostate cancer and low volume skeletal metastases: results of a feasibility and case-control study. The Journal of urology 2015; 193(3): 832-8.
28. Bianchini D, Lorente D, Rescigno P, et al. Effect on Overall Survival of Locoregional Treatment in a Cohort of De Novo Metastatic Prostate Cancer Patients: A Single Institution Retrospective Analysis From the Royal Marsden Hospital. Clinical genitourinary cancer 2017; 15(5): e801-e7.
29. Gratzke C, Engel J, Stief CG. Role of radical prostatectomy in metastatic prostate cancer: data from the Munich Cancer Registry. European urology 2014; 66(3): 602-3.
30. Culp SH, Schellhammer PF, Williams MB. Might men diagnosed with metastatic prostate cancer benefit from definitive treatment of the primary tumor? A SEER-based study. European urology 2014; 65(6): 1058-66.
31. O'Shaughnessy MJ, McBride SM, Vargas HA, et al. A Pilot Study of a Multimodal Treatment Paradigm to Accelerate Drug Evaluations in Early-stage Metastatic Prostate Cancer. Urology 2017; 102: 164-72.
32. Satkunasivam R, Kim AE, Desai M, et al. Radical Prostatectomy or External Beam Radiation Therapy vs No Local Therapy for Survival Benefit in Metastatic Prostate Cancer: A SEER-Medicare Analysis. The Journal of urology 2015; 194(2): 378-85.
33. Loppenberg B, Dalela D, Karabon P, et al. The Impact of Local Treatment on Overall Survival in Patients with Metastatic Prostate Cancer on Diagnosis: A National Cancer Data Base Analysis. European urology 2017; 72(1): 14-9.
34. Boeve LMS, Hulshof M, Vis AN, et al. Effect on Survival of Androgen Deprivation Therapy Alone Compared to Androgen Deprivation Therapy Combined with Concurrent Radiation Therapy to the Prostate in Patients with Primary Bone Metastatic Prostate Cancer in a Prospective Randomised Clinical Trial: Data from the HORRAD Trial. European urology 2019; 75(3): 410-8.
35. Parker CC, James ND, Brawley CD, et al. Radiotherapy to the primary tumour for newly diagnosed, metastatic prostate cancer (STAMPEDE): a randomised controlled phase 3 trial. Lancet (London, England) 2018; 392(10162): 2353-66.
36. Sweeney CJ, Chen Y-H, Carducci M, et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. New England Journal of Medicine 2015; 373(8): 737-46.
37. Burdett S, Boeve LM, Ingleby FC, et al. Prostate Radiotherapy for Metastatic Hormone-sensitive Prostate Cancer: A STOPCAP Systematic Review and Meta-analysis. European urology 2019; 76(1): 115-24.
38. Potters L, Kavanagh B, Galvin JM, et al. American Society for Therapeutic Radiology and Oncology (ASTRO) and American College of Radiology (ACR) practice guideline for the performance of stereotactic body radiation therapy. International journal of radiation oncology, biology, physics 2010; 76(2): 326-32.
39. De Bleser E, Tran PT, Ost P. Radiotherapy as metastasis-directed therapy for oligometastatic prostate cancer. Current opinion in urology 2017; 27(6): 587-95.
40. Decaestecker K, De Meerleer G, Lambert B, et al. Repeated stereotactic body radiotherapy for oligometastatic prostate cancer recurrence. Radiat Oncol 2014; 9: 135-.
41. Palma DA, Salama JK, Lo SS, et al. The oligometastatic state - separating truth from wishful thinking. Nature reviews Clinical oncology 2014; 11(9): 549-57.
42. Riva G, Marvaso G, Augugliaro M, et al. Cytoreductive prostate radiotherapy in oligometastatic prostate cancer: a single centre analysis of toxicity and clinical outcome. Ecancermedicalscience 2017; 11: 786.
43. Ost P, Reynders D, Decaestecker K, et al. Surveillance or Metastasis-Directed Therapy for Oligometastatic Prostate Cancer Recurrence: A Prospective, Randomized, Multicenter Phase II Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018; 36(5): 446-53.
44. Nguyen PL, Alibhai SM, Basaria S, et al. Adverse effects of androgen deprivation therapy and strategies to mitigate them. European urology 2015; 67(5): 825-36.
45. Duchesne GM, Woo HH, Bassett JK, et al. Timing of androgen-deprivation therapy in patients with prostate cancer with a rising PSA (TROG 03.06 and VCOG PR 01-03 [TOAD]): a randomised, multicentre, non-blinded, phase 3 trial. The Lancet Oncology 2016; 17(6): 727-37.
46. Ost P, Bossi A, Decaestecker K, et al. Metastasis-directed therapy of regional and distant recurrences after curative treatment of prostate cancer: a systematic review of the literature. European urology 2015; 67(5): 852-63.
47. Steuber T, Jilg C, Tennstedt P, et al. Standard of Care Versus Metastases-directed Therapy for PET-detected Nodal Oligorecurrent Prostate Cancer Following Multimodality Treatment: A Multi-institutional Case-control Study. European urology focus 2018.
48. Siva S, Bressel M, Murphy DG, et al. Stereotactic Abative Body Radiotherapy (SABR) for Oligometastatic Prostate Cancer: A Prospective Clinical Trial. European urology 2018; 74(4): 455-62.
49. Ploussard G, Gandaglia G, Borgmann H, et al. Salvage Lymph Node Dissection for Nodal Recurrent Prostate Cancer: A Systematic Review. European urology 2018.
50. Zattoni F, Nehra A, Murphy CR, et al. Mid-term Outcomes Following Salvage Lymph Node Dissection for Prostate Cancer Nodal Recurrence Status Post-radical Prostatectomy. European urology focus 2016; 2(5): 522-31.
51. Fossati N, Suardi N, Gandaglia G, et al. Identifying the Optimal Candidate for Salvage Lymph Node Dissection for Nodal Recurrence of Prostate Cancer: Results from a Large, Multi-institutional Analysis. European urology 2019; 75(1): 176-83.
52. Standard Systemic Therapy With or Without Definitive Treatment in Treating Participants With Metastatic Prostate Cancer. 2019. https://clinicaltrials.gov/ct2/show/NCT03678025. (accessed August 8th 2019).
53. Stereotactic Body Radiation for Prostate Oligometastases (ORIOLE). NCT02680587. 2019. https://clinicaltrials.gov/ct2/show/ (accessed August 8th 2019).
54. Radwan N, Phillips R, Ross A, et al. A phase II randomized trial of Observation versus stereotactic ablative RadiatIon for OLigometastatic prostate CancEr (ORIOLE). BMC cancer 2017; 17(1): 453.
55. Rowe SP, Macura KJ, Mena E, et al. PSMA-Based [(18)F]DCFPyL PET/CT Is Superior to Conventional Imaging for Lesion Detection in Patients with Metastatic Prostate Cancer. Mol Imaging Biol 2016; 18(3): 411-9.
56. Sooriakumaran P. Testing radical prostatectomy in men with prostate cancer and oligometastases to the bone: a randomized controlled feasibility trial. BJU international 2017; 120(5b): E8-e20.
Recurrent Urinary Tract Infection (rUTI) in Women Clinical Care Pathway
EVALUATION
Recurrent Urinary Tract Infections (rUTI) is defined as: >2 culture-positive UTIs in 6 months or >3 in one year
1. Take A History - Document symptoms and signs that characterize rUTI episodes and exclude other disorders that could cause the patient’s symptoms (e.g. bacterial vaginosis, vaginal yeast infection, STIs). Ask about the relationship of UTIs to sexual activity, prolonged bladder holding, and bowel irregularity. Rule out noninfectious pelvic and urinary tract sources of symptoms (e.g. overactive bladder, radiation cystitis, bladder pain syndrome, vulvodynia, pelvic organ prolapse, urinary retention/poor bladder emptying, and neurogenic bladder).
Genito-urinary history: Evaluate for complicating factors including pediatric/congenital urologic conditions (e.g., vesicoureteral reflux, megaureter), nephrolithiasis, flank pain, incomplete bladder emptying, prior bladder/pelvic surgery (especially use of mesh material), vaginal pain, cysts or lesions in the vagina, antiestrogen use, pelvic malignancy, absorbent pad use, fecal incontinence, immunosuppression, and neurologic disease.
Review prior urine culture results to confirm rUTI diagnosis with two or more positive cultures in 6 months or 3 or more in one year.
2. Perform Physical Exam to include a pelvic examination - Assess for hygiene and fecal contamination, look for urethral diverticulum, vaginal discharge and/or yeast, vaginal cysts, atrophic vaginitis, and pelvic organ prolapse. If prior mesh or pelvic surgery, consider cystoscopy if a vaginal cyst or periurethral fluctuance (pseudo-urethral abscess formation) order pelvic MRI with coil.
3. Collect Urine Specimen for Testing - Send urine specimen for urinalysis (UA) if symptoms suggestive of UTI. If positive, send the specimen for urine culture and sensitivities. If microscopic hematuria in the absence of infection work-up alongside initial therapy. If sterile pyuria, rule out STIs and urinary TB (AFB’s). If asymptomatic bacteriuria, do not treat. Assess for bacterial persistence, if present considers cross-sectional imaging (retroperitoneal ultrasound) and +/- cystoscopy.
4. Perform ultrasound (bladder scan) US PVR (immediately after voiding) - if >150 cc (on 2 separate occasions) and monitor. Consider intermittent self-catheterization if no other source for infection identified and 1st line suppression fails.
TREATMENT
1st Line Therapy (Behavioral/Lifestyle Modifications) and UTI Prevention Measures- Diabetic patients: Control blood glucose and avoid glucosuria
- Fluid intake: Maintain adequate hydration (Hooten recommended drinking 1.5 L of water daily along with the suggestion to start a 500 mL bottle of water at the beginning of every meal and fully drink it before the next meal).
- Dietary Changes: Cranberry use in all its forms is not supported by current evidence; however, little harm is associated with its use.
- Probiotic: Use of either oral or intravaginal probiotics to restore the natural vaginal microbiota (Lactobacillus spp. colonies) seems to be a promising approach to reducing antibiotic consumption and to decreasing antimicrobial resistance. Lactobacilli may especially be useful for women with histories of recurrent, complicated UTIs or on prolonged antibiotic use. Dietary sources of probiotics may be helpful, these include probiotic yogurt (such as Activia), Greek yogurt, kefir and kombucha juice.
- Voiding habits: Avoid prolonged holding of urine, avoid delaying urination (peeing)
- Hygiene: Avoid disruption of normal vaginal flora with spermicides and/ or harsh cleansers. Avoid soaking in tubs, hot tubs and in baths.
- Sex: Void before and after intercourse and avoid sequential anal and vaginal intercourse
- Bowel Regimen – If diarrhea and/or fecal incontinence are present start loperamide. If mild-moderate constipation is present begin bowel regimen including increasing dietary fiber, stool softener, and/or Miralax. If moderate-severe constipation is present begin bowel regimen (as above) if not on one and consider referral to Primary Care/GI if refractory.
- Persisting irritative bladder symptoms: Many women experience irritative bladder symptoms of pressure or discomfort, bladder urgency, burning when peeing after adequate antibiotic treatment. These symptoms can mimic UTI symptoms and many women are erroneously prescribed another antibiotic but do not exhibit a relief of these irritative symptoms. Consider one of the following treatment regimens:
- Prescribe Urelle 81 mg tab, 4 times a day (evenly spaced). Take each dose with a full glass of water (8 ounces/240 mL). Can take with food if GI upset occurs.
- Over-the-counter medications:
- Azo-Pyridium (phenazopyridine) 100 mg or Uricalm MAX 99.5 mg (phenazopyridine). These are not prescription medications but can be found at most pharmacies or drug stores.
- NSAID (Motrin, Aleve) as the symptoms of UTIs are mostly connected to the inflammatory reaction of the urinary tract due to a significant increase in urinary prostaglandin production, because the onset and duration of clinical symptoms of UTI seem to be strongly connected to prostaglandin levels. Since NSAIDs can inhibit the biosynthesis of prostaglandins, they can be useful in alleviating the symptoms of UTI.
Follow up in 6 months with the maintenance of records of all symptomatic episodes, antibiotic use, UA, and urine culture results. If 2 or more infections in 6 months move to 2nd line therapy.
2nd Line Therapy (Medications)
Review and Reinforce 1st line Therapy +
- In Postmenopausal Women with rUTIs:
- Initiate transvaginal (topical) estrogen and lactobacillus containing probiotics daily. Vaginal estrogen can be prescribed as Estrace cream 1 gram or Premarin cream 0.5 grams, Vagifem (Yuvafem) 10 mcg tablets (all can be used daily for 2 weeks then twice weekly), Estring, or compounded estrogen for approximately $50 a tube.
- Estrace cream comes with 1 applicator that always has to be cleaned. Go on the manufacturer's website (www.estracecream.com), click “Request Applicators” and get 24 individually wrapped applicators at no cost, every 3 months.
- If already on these measures or they are contraindicated or ineffective after 6 months, add methenamine hippurate (Hiprex) 1 gram twice daily plus vitamin C 500 mg twice daily.
- Avoid Vitamin C if history of kidney stones
- If renal insufficiency/decreased GFR, dose methenamine hippurate accordingly
- In severe cases, consider combined initial therapy with vaginal estrogen, probiotics, methenamine hippurate and vitamin C.
- An alternative to methenamine hippurate is d-Mannose 500- 750 mg twice/day, a monosaccharide that can be rapidly absorbed and excreted by the urinary tract and can prevent the adhesion of type 1 bacterial fimbriae (bacterial virulence factor, usually caused by E. coli) promoting UTI to the uroepithelium
- In Premenopausal Women with rUTIs
- With Post-Coital rUTIs:
- Initiate a single low dose prophylactic antibiotic* within 2 hours of sexual activity for 6-12 months duration OR
- Has already tried post-coital antibiotics, or has intercourse more than twice per week initiate methenamine hippurate 1 gram twice daily, vitamin C 500 mg twice daily and lactobacillus containing probiotics daily or alternative to methenamine hippurate is d-Mannose 500- 750 mg twice/day.
- With Post-Coital rUTIs:
- With rUTI unrelated to sexual activity:
- Consider daily low dose prophylactic antibiotic* for 6-12 months’ duration OR
- Have already tried daily antibiotics, initiate methenamine hippurate 1 gram twice daily, vitamin C 500 mg twice daily and lactobacillus containing probiotics daily or alternative to methenamine hippurate is d-Mannose 500- 750 mg twice/day
Follow up in 6 months with the maintenance of records of all symptomatic episodes, antibiotic use, UA, and urine culture results. If refractory; consider an evaluation with cross sectional imaging +/- cystoscopy. In refractory patients, alternative prophylactic antibiotics can be tried. In severe refractory cases in patients on intermittent catheterization, intravesical gentamycin can be considered.
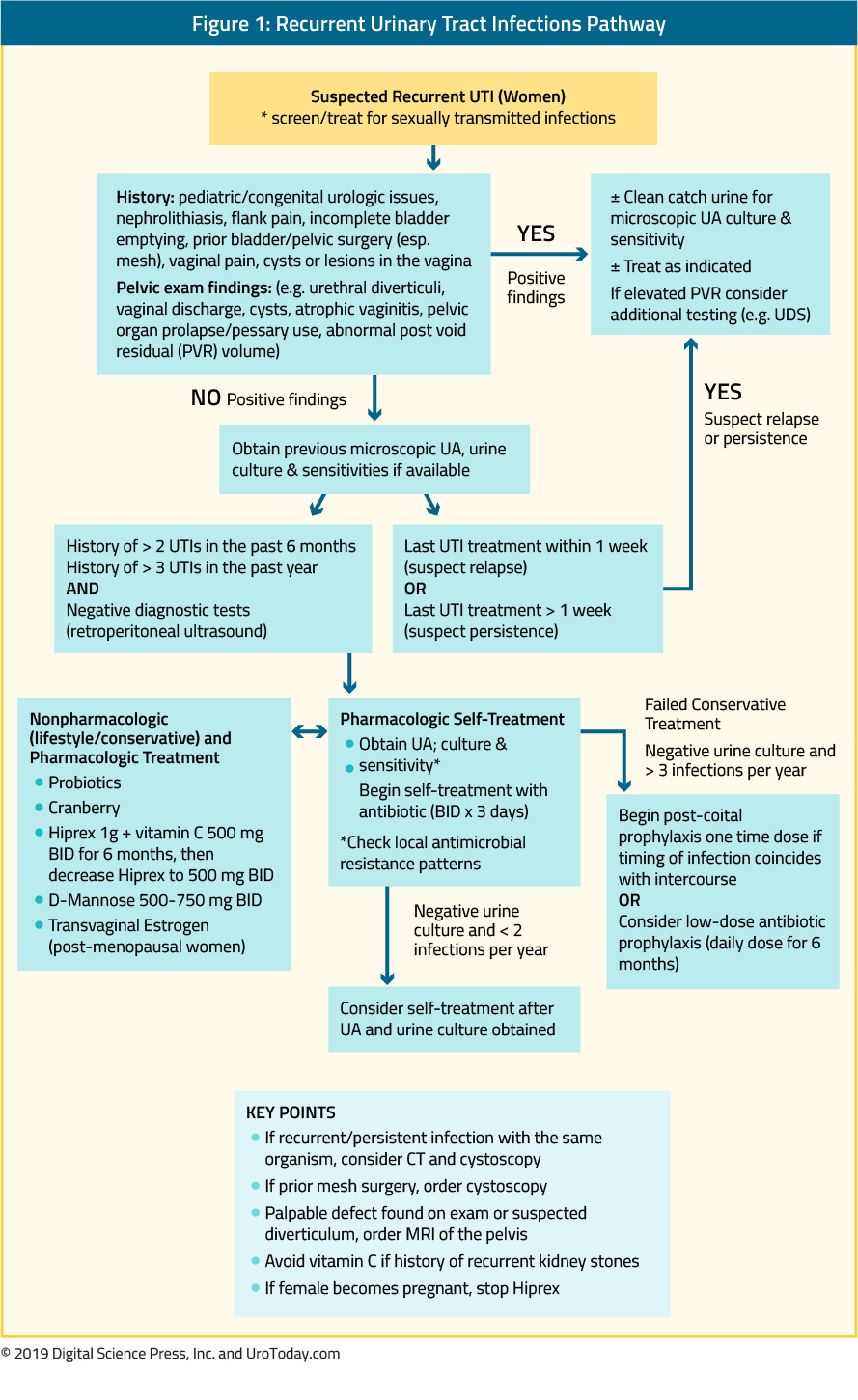
Published Date: September 26th, 2019
- Written by: Diane K. Newman, DNP, ANP-BC, FAAN
- References:
1. American Urological Association, the Canadian Urological Association and the Society of Urodynamics Female Pelvic Medicine and Urogenital Reconstruction. Recurrent uncomplicated UTIs in women.
2. Hooton TM, Vecchio M, Iroz A, Tack I, Dornic Q, Seksek I, Lotan Y. Effect of increased daily water intake in premenopausal women with recurrent urinary tract infections: A randomized clinical trial. JAMA Intern Med. 2018 Nov 1;178(11):1509-1515. doi: 10.1001/jamainternmed.2018.4204
3. Nicolle et al., Practice Guideline for the Management of Asymptomatic Bacteriuria: 2019 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2019 Mar 21. pii: ciy1121. doi: 10.1093/cid/ciy1121).
4. Smith AL, Brown J, Wyman JF, Berry A, Newman DK, Stapleton AE. Treatment and prevention of recurrent lower urinary tract infections in women: A rapid review with practice recommendations. J Urol. 2018 Dec;200(6):1174-1191. doi: 10.1016/j.juro.2018.04.088).
5. Wawrysiuk S, Naber K, Rechberger T, Miotla P. Prevention and treatment of uncomplicated lower urinary tract infections in the era of increasing antimicrobial resistance-non-antibiotic approaches: a systemic review. Arch Gynecol Obstet. 2019 Jul 26. doi: 10.1007/s00404-019-05256-
Prostate Cancer Survivorship
Physical Side Effects
Urinary Dysfunction
Urinary dysfunction is a side effect of both surgical and radiotherapy (RT) for local treatment of prostate cancer (PCa). Surgical side effects typically include a period of urinary incontinence for several months postoperatively followed by a degree of stress urinary incontinence that may persist for months or even years. RT-induced urinary dysfunction typically manifests as bladder irritability/overactivity either during treatment or shortly thereafter. Longer-term urinary dysfunction issues after RT may include urethral strictures necessitating periodic interventions and/or catheterization.
The ProtecT trial randomized 1,643 men from 1999 to 2009 to undergoing either active monitoring (n=545), surgery (n=553), or RT (n=545), finding that at a median 10 years of follow-up, PCa-specific mortality was low irrespective of treatment.2 As part of this trial, patient-reported outcomes were collected and have now become one of the benchmarks for counseling patients with regards to long-term side effects of treatment for localized PCa treatment.3 Questionnaires were completed at the time of diagnosis, at 6 and 12 months after randomization, and annually thereafter. Patients completed validated measures that assessed urinary, bowel, and sexual function and specific effects on quality of life, anxiety, and depression, and general health. The rate of questionnaire completion during follow-up was outstanding at >85% for most measures. Regarding urinary dysfunction, radical prostatectomy (RP) had the greatest negative effect on urinary continence, and although there was some recovery over time, these patients remained worse throughout follow-up compared to patients undergoing active monitoring or RT. Interestingly, RT had little effect on urinary incontinence, and there was a gradual decrease in urinary function over time for the men undergoing active monitoring. Urinary voiding and nocturia were worse in the radiotherapy group at 6 months but then mostly recovered and were similar to the other groups after 12 months. Urinary incontinence has been cited as being the most important factor for decision regret among receiving local therapy for PCa and may be incompletely explained/discussed with ~80% of patients prior to undergoing treatment.4
Sexual Dysfunction
Similar to urinary dysfunction, sexual dysfunction is a common side effect of localized therapy for PCa. Patients undergoing RP will suffer a degree of sexual dysfunction in the immediate postoperative period with a degree of recovering over 12-24 months after surgery. Many studies have been published assessing predictors of postoperative recovery of sexual function, commonly highlighting younger age and adequate function pre-operatively as predictors of post-operative recovery. Men undergoing RT, similar to urinary dysfunction, will not notice an immediate effect on sexual function during the treatment phase, but generally, suffer sexual dysfunction in the years post-radiation.
In the ProtecT trial, RP incurred the greatest degree of sexual dysfunction among all three treatment arms, with some recovery of function over time.3 The negative effect of RT on sexual function was greatest at 6 months, but sexual function then recovered somewhat and was stable thereafter. Sexual dysfunction also declined in the active monitoring group over time.
Primarily secondary to the sexual side effects of localized treatment for PCa, many cancer centers now have fellowship-trained experts that see these patients concomitantly with the oncologist. There are a variety of treatment options offered, including oral PDE-5 inhibitors (sildenafil, tadalafil, etc.), intracavernosal injection therapy, and penile prosthetics.
Bowel Dysfunction
Bowel dysfunction is typically low for patients undergoing RP or active surveillance (AS) but may be a detrimental side effect among men undergoing RT. In the ProtecT trial, bowel function was worse in the RT group at 6 months than in the other groups but then recovered somewhat, except for the increasing frequency of bloody stools; bowel function was unchanged in the active monitoring and RP groups.3
Bowel dysfunction and rectal toxicity has improved with the recent FDA approval of hydrogel rectal spacers. Prior to RT, patients may have a hydrogel rectal spacer (SpaceOAR®) placed in a transperineal fashion in the fat between the rectum and Denonvilliers' fascia. In the pivotal clinical trial assessing hydrogel spacers, 114 patients were enrolled between 2010 and 2011 with 54 patients selected for a hydrogel injection before the beginning of RT.5 Patients were surveyed at various time-points with the EPIC PCa questionnaire – among patients treated with a hydrogel spacer, mean bowel function and bother score changes of >5 points in comparison with baseline levels were found only at the end of RT (10-15 points; p < 0.01). Mean bowel bother score changes of 21 points at the end of RT, 8 points at 2 months, 7 points at 17 months, and 6 points at 63 months after RT were found for patients treated without a spacer. These bowel quality of life results have given hydrogel spacers an option among patients considering RT.
Other health-related effects
There is evidence that both RT and androgen deprivation therapy (ADT) may contribute to the development of coronary heart disease, sudden cardiac death, myocardial infarction, and skeletal-related events such as fracture.6
Psychological Side Effects
Depression and AnxietyDepression is the most common psychiatric comorbidity among cancer patients, including patients with PCa. Ravi et al.7 previously utilized the SEER-Medicare database to assess the burden of mental health issues (anxiety, major depressive disorder, suicide) in patients with localized PCa. Among 50,586 men >65 years of age without a diagnosis of mental illness, 20.4% of men developed a mental illness with a median 55-month follow-up. Interestingly, patients undergoing WW (29.7%) and RT (29.0%) had a significantly increased incidence of mental illness compared to patients undergoing RP (22.6%; p<0.001). A systematic review of depression and anxiety in patients with PCa identified 27 articles comprising 4,494 patients.8 The meta-analysis of prevalence rates identified pretreatment prevalence of depression of 17.27% (95% confidence interval (CI) 15.06%-19.72%), on-treatment prevalence of 14.70% (95% CI 15.06%-19.72%) and post-treatment prevalence of 18.44% (95% CI 15.18%-22.22%). For anxiety, pretreatment prevalence was 27.04% (95% CI 24.26%-30.01%), on-treatment was 15.09% (95% CI 12.15%-18.60%) and post-treatment was 18.49% (95% CI 13.81%-24.31%). For patients undergoing AS, nearly one-third of patients (29%) report cancer-specific anxiety in the year following diagnosis.9 Interestingly, over time, this anxiety decreased significantly.
There is also increasing evidence that ADT for locally advanced and metastatic PCa is associated with depression. A study from 2016 using SEER-Medicare data found that men that received ADT, compared with patients who did not receive ADT, had higher 3-year cumulative incidences of depression (7.1% v 5.2), inpatient psychiatric treatment (2.8% v 1.9%), and outpatient psychiatric treatment (3.4% v 2.5%).10 Furthermore, the risk of depression increased with the duration of ADT, from 12% with ≤ 6 months of treatment, 26% with 7 to 11 months of treatment, to 37% with ≥ 12 months of treatment. A recent meta-analysis of 18 studies among 168,756 men found that ADT use conferred a 41% increased risk of depression (RR 1.41, 95%CI 1.18-1.70).11 These results were consistent when limiting the analysis to studies in localized disease (relative risk (RR) 1.85, 95%CI 1.20-2.85). Interestingly, this analysis did not find an association for continuous ADT with depression risk compared to intermittent ADT (RR 1.00, 95%CI 0.50-1.99).
Suicidal Risk
Patients with PCa have been shown to be at increased risk of suicide across several population-level studies. In a SEER analysis assessing suicide risk among patients with genitourinary malignancies from 1988-2010, Klaassen et al.12 found an age-adjusted standardized mortality ratio (SMR) of 1.37 for patients with PCa (95%CI, 0.99-1.86) Increasing age, metastatic disease and Caucasian race were risk factors for suicide among these patients. Interestingly, even patients >15 years after diagnosis were at increased risk of suicide compared to the general population (SMR 1.84, 95%CI 1.39-2.41). In an assessment of PCa suicidal risk compared to individuals with other malignancies, Dalela et al.13 found that risk of suicidal death was no different in men with PCa (1,165 [0.2%]) compared to men with other cancers (2,232 [0.2%]), However, within the first year of diagnosis, men with PCa had an increased risk of suicide (absolute risk reduction (ARR) 3.98, 95% CI 3.02-5.23 0-3 months after diagnosis). Furthermore, men with non-metastatic PCa who were Caucasian, uninsured, or recommended but did not receive treatment (hazard ratio (HR) vs treated 1.44, 95%CI 1.20-1.72) were at increased risk of suicidal death.
A meta-analysis of observational studies assessing incidence and risk factors of suicide after PCa diagnosis was recently published.14 This study included 8 observational studies involving 1,281,393 men diagnosed with PCa and 842,294 matched PCa-free men. Guo et al. found an overall increased relative risk of suicide of 2.01 (95% CI 1.52-2.64) among men diagnosed with PCa compared with those without PCa during the first year after diagnosis, particularly during the first 6 months after diagnosis (RR 2.24, 95%CI 1.77-2.85). Additionally, PCa patients were at an increased risk of suicide among men aged 75 years or older (RR 1.51, 95% CI 1.04-2.18) and for those treated with ADT (RR 1.80, 95% CI 1.54-2.12).
Until recently, all population-level studies assessing risk of suicide among PCa patients have not accounted for psychiatric comorbidities at the time of diagnosis. This is important, considering that being unable to adjust for psychiatric comorbidities makes it impossible to assess the true risk associated with a PCa diagnosis on suicidal risk. At the AUA 2019 annual meeting, Klaassen et al.15 presented data assessing all residents of Ontario, Canada diagnosed with either prostate, bladder or kidney cancer (1997-2014). Each patient was assigned a psychiatric utilization gradient (PUG) score in the five years prior to cancer diagnosis: 0 (none), 1 (outpatient), 2 (emergency department), 3 (hospital admission). Non-cancer controls were matched 4:1 to cancer patients based on sociodemographic variables and a marginal cause-specific hazard model was used to assess the effect of cancer on the risk of suicidal death. Among 191,068 patients included (137,699 PCa, 29,884 bladder cancer, 23,485 kidney cancer), 109,154 (57.1%) were PUG score 0, 79,553 (41.6%) PUG score 1, 1,596 (0.84%) PUG score 2, and 765 (0.40%) PUG score 3. Patients with genitourinary cancer had a higher risk of dying of suicide compared to controls (HR 1.16, 95%CI 1.00-1.36). Specifically, among individuals with PUG score 0, those with cancer were significantly more likely to die of suicide compared to patients without cancer (HR 1.39, 95%CI 1.12-1.74).
Guideline Recommendations
The Commission on Cancer requires cancer programs to develop and implement processes to monitor formation and dissemination of a survivorship care plan for all cancer patients with stage I-III disease treated with curative intent, and to have this plan in place within 1-year of diagnosis of cancer and no later than 6 months after completing adjuvant therapy.16 Guideline recommendations for PCa survivorship have primarily been driven by the American Cancer Society (ACS) and the American Society of Clinical Oncology (ASCO). The ACS noted in their 2014 guideline that survivorship should promote comprehensive follow-up care and optimal health and quality of life for the post-treatment PCa survivor.17 The guidelines also address health promotion, surveillance for PCa recurrence, screening for second primary cancers, long-term and late effects assessment and management, psychosocial issues, and care coordination among the oncology team, primary care clinicians, and non-oncology specialists. Subsequently, the ASCO Endorsement Panel reviewed the ACS guidelines, endorsing these guidelines with the following recommendations:18• Measure PSA level every 6 to 12 months for the first 5 years and then annually, considering more frequent evaluation in men at high risk for recurrence and in candidates for salvage therapy.
• Refer survivors with elevated or increasing PSA levels back to their primary treating physician for evaluation and management.
• Adhere to ACS guidelines for the early detection of cancer.
• Assess and manage physical and psychosocial effects of PCa and its treatment.
• Annually assess for the presence of long-term or late effects of PCa and its treatment.
Screening Measures
There are several screening tools to assess for quality of life, depression and suicidal risk. A study from 2017 assessed differences in the scores, relative severity and major depressive disorder from three standardized self-report scales for depression in PCa patients [The Hospital Anxiety and Depression Scale Depression subscale (HADS-D), the Self-rating Depression Scale (SDS) and the Patient Health Questionnaire (PHQ-9) for depression].19 Among 138 PCa patients, despite significant correlations between the total scores from the three scales, severity classification differed across the three scales. Furthermore, there was considerable underestimation of depression by the HADS-D compared to the PHQ-9 and a similar tendency for the SDS. This study highlights that scale construction and depression items included can produce different results across scales, making inter-study comparisons difficult. Despite these findings, we recommend that at minimum oncologists should be using at least one depression index to assess patient well-being at each clinic visit.In addition to the aforementioned HADS-D, SDS, and PHQ-9 metrics, the National Comprehensive Cancer Network (NCCN) provides a guideline for identifying and explaining risk factors in patients with cancer, in addition to providing a “distress thermometer”. The NCCN defines distress, in the setting of cancer, as a multifactorial emotional experience of a psychological, social, and/or spiritual nature that may interfere with the ability to cope effectively with the diagnosis.20 Distress can range from sadness and fear to more disabling symptoms such as anxiety and depression. Furthermore, the time periods at which patients are at increased vulnerability begin with the realization of a suspicious symptom, all the way through to failure/disease recurrence and near the end of life. The NCCN recommends screening all patients for distress to recognize, monitor, and treat patients effectively.20
Previous work has also suggested that screening for depression and erectile dysfunction may be a way to decrease suicidal risk among PCa patients.21 A proposed algorithm allows for an initial evaluation with the EPIC-CP and PHQ-9 tools to assess for health-related quality of life and depression, respectively. If the EPIC-CP or PHQ-9 are negative for depression or erectile dysfunction, these tools should still be used at each visit to regularly evaluate patients. If EPIC-CP or PHQ-9 suggest problems with depression or erectile dysfunction, then an 8-question suicidal ideation questionnaire (adapted from Recklitis et al.22) should be completed. If the suicidal ideation questionnaire demonstrates any level of suicidal ideation, clinicians should make an urgent referral for psychiatric evaluation. This is particularly true when the patient has the concomitant high-risk suicidal risk profile of being elderly, white, single, or with high-risk or disease progression. Given that, at maximum, the patient must answer a 27-point composite questionnaire, this should be feasible in the busy clinical setting and can be provided to the patient at appointment check-in and completed in the waiting room before the physician-patient encounter. Regardless of the results from these screening tools, if any member of the healthcare team has an index of suspicion for suicidal ideation, the physician should immediately make a referral for psychiatric evaluation.
Conclusions
With nearly 3 million men in the United States living with PCa, survivorship programs are now mandated by the Commission on Cancer and play an integral role in health and well-being of men with PCa. In addition to the physical side effects of treatment that should be addressed at each clinic visit, there are crucial psychiatric side effects, including depression, anxiety, and suicidal ideation that should be screened for and recognized by all members of the healthcare team.Published Date: December 2019
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34.
2. Hamdy FC, Donovan JL, Lane JA, et al. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. N Engl J Med. 2016;375(15):1415-1424.
3. Donovan JL, Hamdy FC, Lane JA, et al. Patient-Reported Outcomes after Monitoring, Surgery, or Radiotherapy for Prostate Cancer. N Engl J Med. 2016;375(15):1425-1437.
4. Albkri A, Girier D, Mestre A, Costa P, Droupy S, Chevrot A. Urinary Incontinence, Patient Satisfaction, and Decisional Regret after Prostate Cancer Treatment: A French National Study. Urol Int. 2018;100(1):50-56.
5. Pinkawa M, Berneking V, Schlenter M, Krenkel B, Eble MJ. Quality of Life After Radiation Therapy for Prostate Cancer With a Hydrogel Spacer: 5-Year Results. Int J Radiat Oncol Biol Phys. 2017;99(2):374-377.
6. Wallis CJ, Mahar AL, Satkunasivam R, et al. Cardiovascular and Skeletal-related Events Following Localized Prostate Cancer Treatment: Role of Surgery, Radiotherapy, and Androgen Deprivation. Urology. 2016;97:145-152.
7. Ravi P, Karakiewicz PI, Roghmann F, et al. Mental health outcomes in elderly men with prostate cancer. Urol Oncol. 2014;32(8):1333-1340.
8. Watts S, Leydon G, Birch B, et al. Depression and anxiety in prostate cancer: a systematic review and meta-analysis of prevalence rates. BMJ Open. 2014;4(3):e003901.
9. Marzouk K, Assel M, Ehdaie B, Vickers A. Long-Term Cancer Specific Anxiety in Men Undergoing Active Surveillance of Prostate Cancer: Findings from a Large Prospective Cohort. J Urol. 2018;200(6):1250-1255.
10. Dinh KT, Reznor G, Muralidhar V, et al. Association of Androgen Deprivation Therapy With Depression in Localized Prostate Cancer. J Clin Oncol. 2016;34(16):1905-1912.
11. Nead KT, Sinha S, Yang DD, Nguyen PL. Association of androgen deprivation therapy and depression in the treatment of prostate cancer: A systematic review and meta-analysis. Urol Oncol. 2017;35(11):664 e661-664 e669.
12. Klaassen Z, Jen RP, DiBianco JM, et al. Factors associated with suicide in patients with genitourinary malignancies. Cancer. 2015;121(11):1864-1872.
13. Dalela D, Krishna N, Okwara J, et al. Suicide and accidental deaths among patients with non-metastatic prostate cancer. BJU Int. 2016;118(2):286-297.
14. Guo Z, Gan S, Li Y, et al. Incidence and risk factors of suicide after a prostate cancer diagnosis: a meta-analysis of observational studies. Prostate Cancer Prostatic Dis. 2018;21(4):499-508.
15. Klaassen Z, Wallis CJ, Goldberg H, et al. Utilization of Psychiatric Resources Prior to Genitourinary (GU) Cancer Diagnosis: Implications for Survival Outcomes. AUA 2019. 2019.
16. Fashoyin-Aje LA, Martinez KA, Dy SM. New patient-centered care standards from the commission on cancer: opportunities and challenges. J Support Oncol. 2012;10(3):107-111.
17. Skolarus TA, Wolf AM, Erb NL, et al. American Cancer Society prostate cancer survivorship care guidelines. CA Cancer J Clin. 2014;64(4):225-249.
18. Resnick MJ, Lacchetti C, Bergman J, et al. Prostate cancer survivorship care guideline: American Society of Clinical Oncology Clinical Practice Guideline endorsement. J Clin Oncol. 2015;33(9):1078-1085.
19. Sharpley CF, Bitsika V, Christie DR, Hunter MS. Measuring depression in prostate cancer patients: does the scale used make a difference? Eur J Cancer Care (Engl). 2017;26(1).
20. National Comprehensive Cancer N. Distress management. Clinical practice guidelines. J Natl Compr Canc Netw. 2003;1(3):344-374.
21. Klaassen Z, Arora K, Wilson SN, et al. Decreasing suicide risk among patients with prostate cancer: Implications for depression, erectile dysfunction, and suicidal ideation screening. Urol Oncol. 2018;36(2):60-66.
22. Recklitis CJ, Zhou ES, Zwemer EK, Hu JC, Kantoff PW. Suicidal ideation in prostate cancer survivors: understanding the role of physical and psychological health outcomes. Cancer. 2014;120(21):3393-3400.
Mapping Progress in Bladder Cancer
- Written by: Ashish Kamat, MD, MBBS
- References:
- National Cancer Institute. Cancer Stat Facts: Bladder Cancer. https://seer.cancer.gov/statfacts/html/urinb.html Accessed January 15, 2019.
- Hermans TJN, Voskuilen CS, van der Heijden MS, et al. Neoadjuvant treatment for muscle-invasive bladder cancer: The past, the present, and the future. Urol Oncol 2018 Sep;36(9):413-422.
- Kamat AM et al. BCG-unresponsive non-muscle-invasive bladder cancer: recommendations from the IBCG. Nat Rev Urol. 2017 Apr;14(4):244-255.
- Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 2017 Mar;376(11):1015-1026.
- Patel MR, Ellerton J, Infante JR, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol 2018 Jan;19(1):51-64.
- Powles T, O'Donnell PH, Massard C, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: Updated results from a phase 1/2 open-label study. JAMA Oncol 2017;3(9):e172411.
- Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016 May 7;387(10031):1909-1920.
- Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. TLancet Oncol 2017 Mar;18(3):312-322.
- Rosenberg JE, Sridhar SS, Zhang J, et al. Updated results from the enfortumab vedotin phase 1 (EV-101) study in patients with metastatic urothelial cancer (mUC). J Clin Oncol 2018 May;36(15_suppl):4504-4504.
- Siefker-Radtke AO, Necchi A, Park SH, et al. First results from the primary analysis population of the phase 2 study of erdafitinib (ERDA; JNJ-42756493) in patients (pts) with metastatic or unresectable urothelial carcinoma (mUC) and FGFR alterations (FGFRalt). J Clin Oncol 2018 May;36(15_suppl):4503-4503.
- Hahn, NM. A Golden Age of Bladder Cancer Drug Development. Urotoday.com
Updates in Systemic Therapy for Upper Tract Urothelial Carcinoma: Perioperative Considerations
Until recently, there have been very limited randomized data to guide the provision of perioperative systemic therapy in upper tract urothelial carcinoma. Thus, data has been extrapolated from patients with urothelial carcinoma of the bladder.
Neoadjuvant therapy
Data for the use of neoadjuvant chemotherapy prior to radical cystectomy in urothelial carcinoma of the bladder are robust: the Advanced Bladder Cancer meta-analysis collaboration, in a meta-analysis of 11 trials with 3,005 patients, demonstrated a significant survival benefit to the use of neoadjuvant platinum-based combination chemotherapy as compared to upfront surgery (hazard ratio 0.86, 95% confidence interval 0.77 to 0.95). In absolute terms, this corresponds to a 5% absolute survival at 5 years.5In contrast, there is extremely limited data regarding the use of neoadjuvant chemotherapy in patients with upper tract urothelial carcinoma. No institution has published data on more than 50 patients who received neoadjuvant chemotherapy prior to nephroureterectomy. In two publications, the MD Anderson Cancer Center group has reported on their experience. Matin et al. reported on 43 patients with biopsy-proven high-grade upper tract urothelial carcinoma who underwent neoadjuvant chemotherapy prior to nephroureterectomy.6 They demonstrated a 14% pathologic complete response rate in this cohort. Additionally, when compared to historical controls, there was an increased likelihood of downstage (p=0.004): pT2 disease was found in 49% compared with 65% in controls and pT3 disease was found in 28% compared with 48%. In a more recent analysis, Porten et al. provided a retrospective review of 31 patients who underwent neoadjuvant chemotherapy prior to nephroureterectomy compared to 81 historical controls who received initial surgery for high-risk upper tract urothelial carcinoma.7 They demonstrated that patients receiving neoadjuvant chemotherapy had improved five year overall survival (80% versus 58%, p=0.02) and five year disease-specific survival (90% versus 58%, p=0.002). After adjusting for relevant patient, treatment and tumor characteristics, the risk of overall mortality (hazard ratio 0.42, p=0.035) and disease-specific mortality (hazard ratio 0.19, p=0.006) were significantly lower among patients who received neoadjuvant therapy.
More recently, Kubota and colleagues reported a multi-institutional analysis of 101 patients with high-risk upper tract urothelial carcinoma (stage cT3-4 or cN+) who received neoadjuvant chemotherapy prior to nephroureterectomy at 5 medical centers in Japan and compared these to 133 patients who received surgery alone.8 Most patients who received neoadjuvant chemotherapy in this cohort were given gemcitabine and carboplatin (75%) while a minority received gemcitabine and cisplatin (21%). The authors found that while neoadjuvant chemotherapy increased pathological downstaging and was independently predictive of improved recurrence-free survival and cancer-specific survival, they were unable to demonstrate improvements in overall survival.
Finally, in 2018, Liao et al reported their experience from Johns Hopkins.9 They compared 32 patients who received neoadjuvant chemotherapy prior to nephroureterectomy for biopsy-proven high-grade upper tract urothelial carcinoma to 208 patients who underwent surgery alone. These authors found a 9.4% pathologic complete response rate in addition to significant pathologic downstaging.
In comparison to the use of neoadjuvant chemotherapy for urothelial carcinoma of the bladder, there is potentially even greater rationale for its use in upper tract urothelial carcinoma. First, patients are much more likely to be able to tolerate a highly efficacious regime (gemcitabine and cisplatin) with two functioning kidneys prior to nephroureterectomy than they will post-operatively. Additionally, important prognostic information can be derived if patients experience pathological downstaging.1 However, one of the primary barriers to the use of neoadjuvant chemotherapy is the lack of reliable pre-operative pathologic specimens to identify invasive disease, as mentioned previously. Second, there are concerns regarding the delay to definitive surgical treatment, particularly in patients who may have chemoresistant disease. Third, there are concerns regarding a potential increase in perioperative morbidity.
Finally, there is a concern of overtreatment through the use of toxic medications in patients who may or may not have invasive disease. There have been attempts to assess the role of neoadjuvant chemotherapy in this patient population. A trial from the University of Michigan was terminated due to poor accrual (NCT01663285). Memorial Sloan Kettering is a sponsor for a trial of gemcitabine and cisplatin as neoadjuvant chemotherapy in patients with upper tract urothelial carcinoma, which according to the last update remains active but is no longer recruiting. This trial sought to enroll 57 participants in a Phase II design to assess the primary outcome of pathological response rate. Additionally, a Phase II ECOG-ACRIN trial seeking to assess pathologic complete response rates following neoadjuvant chemotherapy (MVAC or gemcitabine and cisplatin) among 36 patients with high-grade upper tract urothelial carcinoma. Results have not yet been published though they were presented at AUA 2018. While the gemcitabine and cisplatin arm did not meet accrual, 30 patients were accrued to the ddMVAC arm. Pathologic complete response was seen in 6 of 29 evaluable patients. None of these trials have provided a randomized comparison to surgery alone. In contrast, NCT02876861 is a trial from Xiangya Hospital of Central South University which is actively recruiting patients and randomizing to neoadjuvant chemotherapy with gemcitabine and cisplatin prior to nephroureterectomy or surgery alone. They are targeting accrual of 50 patients to assess the primary outcome of disease-free survival.
Adjuvant chemotherapy
Again, there is strong evidence for adjuvant chemotherapy in urothelial carcinoma of the bladder. In contrast to neoadjuvant chemotherapy, the data for adjuvant chemotherapy in upper tract urothelial carcinoma are more robust.First, there is a meta-analysis of nine retrospective cohort studies comparing 482 patients who received adjuvant chemotherapy to 1,300 patients who were treated with surgery alone. Those who received adjuvant therapy had significantly improved disease-free survival (hazard ratio 0.49, 95% confidence interval 0.24 to 0.99) and overall survival (hazard ratio 0.43, 95% confidence interval 0.21 to 0.89)10. This analysis is limited by significant selection biases.
In contrast, a Phase III randomized trial of perioperative chemotherapy versus surveillance in upper tract urothelial cancer (POUT) is a Phase III, multicenter trial from the Institute of Cancer Research in the United Kingdom which randomized patients who are chemotherapy eligible with pT2-4, pN0-3 or pT1, pN+ following nephroureterectomy to adjuvant platinum-based chemotherapy or surveillance. The results of this trial remain to be published, although they have been presented at ASCO GU and EAU in 2018. To summarize, 123 patients were randomized to surveillance and 125 to adjuvant chemotherapy at 57 different centers. Patients in the intervention arm received either gemcitabine and cisplatin or gemcitabine and carboplatin as their renal function allowed. The majority of enrolled patients had pT3 disease (65%) and were node-negative (pN0, 91%). The authors demonstrated that adjuvant chemotherapy significantly improved disease-free survival (hazard ratio 0.49, 95% confidence interval 0.31 to 0.76) as well as metastasis-free survival (hazard ratio 0.49, 95% confidence interval 0.30 to 0.79). However, overall survival data were not yet mature at the time of presentation, so despite the separation of the curves, a meaningful benefit cannot be shown yet. Grade 3 or greater toxicity was experienced by 60% of patients receiving adjuvant chemotherapy and 24% of patients undergoing surveillance.
Previous analyses have demonstrated that patients are much less likely to derive benefit from non-cisplatin based regimes10. Thus, a significant decline in renal function attributable to nephroureterectomy may result in many patients being unable to receive optimal systemic therapy.
Immunotherapy
The data discussed this far center on the use of cytotoxic chemotherapy, whether gemcitabine and cisplatin, MVAC, or gemcitabine and carboplatin, as these have been the traditional agents used for patients with urothelial carcinoma. Following data supporting the role of immunotherapy using checkpoint inhibitors in patients with advanced metastatic urothelial carcinoma, the recently published Phase II PURE-01 trial examined neoadjuvant pembrolizumab for muscle-invasive urothelial bladder carcinoma11. This analysis demonstrated higher rates of pathologic complete response (42%) and downstaging to non-muscle invasive disease (54%) than would be expected from historical use of cytotoxic chemotherapy, though a randomized control was not employed in this analysis. Analyses of durvalumab plus tremelimumab and of gemcitabine plus cisplatin plus pembrolizumab demonstrated similarly promising results. This approach has not, to our knowledge, been tested in upper tract urothelial carcinoma but such an approach may be particularly valuable in these patients.Conclusions
At the 2018 Society of Urologic Oncology meeting, Dr. Jean Hoffman-Censits presented on the role of neoadjuvant chemotherapy in these patients. She discussed differences between urothelial carcinomas of the bladder and of the upper urinary tract. Notably, she highlighted that pathologic response rates appear to be lower in patients with upper tract urothelial carcinoma (9-15%) as compared to bladder cancer (>20%). The ongoing URANUS trial will better delineate the role of neoadjuvant therapy in upper tract urothelial carcinoma.Published Date: December 2019
- Written by: Zachary Klaassen, MD, MSc
- References:
1. Leow JJ, Chong KT, Chang SL, et al. Upper tract urothelial carcinoma: a different disease entity in terms of management. ESMO Open 2016; 1(6):e000126.
2. Catto JW, Azzouzi AR, Amira N, et al. Distinct patterns of microsatellite instability are seen in tumours of the urinary tract. Oncogene 2003; 22(54):8699-706.
3. Kunze E, Wendt M, Schlott T. Promoter hypermethylation of the 14-3-3 sigma, SYK and CAGE-1 genes is related to the various phenotypes of urinary bladder carcinomas and associated with progression of transitional cell carcinomas. Int J Mol Med 2006; 18(4):547-57.
4. Margulis V, Shariat SF, Matin SF, et al. Outcomes of radical nephroureterectomy: a series from the Upper Tract Urothelial Carcinoma Collaboration. Cancer 2009; 115(6):1224-33.
5. Advanced Bladder Cancer Meta-analysis C. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data advanced bladder cancer (ABC) meta-analysis collaboration. Eur Urol 2005; 48(2):202-5; discussion 205-6.
6. Matin SF, Margulis V, Kamat A, et al. Incidence of downstaging and complete remission after neoadjuvant chemotherapy for high-risk upper tract transitional cell carcinoma. Cancer 2010; 116(13):3127-34.
7. Porten S, Siefker-Radtke AO, Xiao L, et al. Neoadjuvant chemotherapy improves survival of patients with upper tract urothelial carcinoma. Cancer 2014; 120(12):1794-9.
8. Kubota Y, Hatakeyama S, Tanaka T, et al. Oncological outcomes of neoadjuvant chemotherapy in patients with locally advanced upper tract urothelial carcinoma: a multicenter study. Oncotarget 2017; 8(60):101500-101508.
9. Liao RS, Gupta M, Schwen ZR, et al. Comparison of Pathological Stage in Patients Treated with and without Neoadjuvant Chemotherapy for High Risk Upper Tract Urothelial Carcinoma. J Urol 2018; 200(1):68-73.
10. Leow JJ, Martin-Doyle W, Fay AP, et al. A systematic review and meta-analysis of adjuvant and neoadjuvant chemotherapy for upper tract urothelial carcinoma. Eur Urol 2014; 66(3):529-41.
11. Necchi A, Anichini A, Raggi D, et al. Pembrolizumab as Neoadjuvant Therapy Before Radical Cystectomy in Patients With Muscle-Invasive Urothelial Bladder Carcinoma (PURE-01): An Open-Label, Single-Arm, Phase II Study. J Clin Oncol 2018:JCO1801148.
A Golden Age of Bladder Cancer Drug Development
- Written by: Noah M. Hahn, MD
- References:
- Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507:315-22, 2014
- Bellmunt J, de Wit R, Vaughn DJ, et al: Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med 376:1015-1026, 2017
- Patel MR, Ellerton J, Infante JR, et al: Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol 19:51-64, 2018
- Powles T, O'Donnell PH, Massard C, et al: Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: Updated results from a phase 1/2 open-label study. JAMA Oncology 3:e172411, 2017
- Rosenberg JE, Hoffman-Censits J, Powles T, et al: Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. The Lancet 387:1909-1920, 2016
- Sharma P, Retz M, Siefker-Radtke A, et al: Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. The Lancet Oncology 18:312-322, 2017
- Rosenberg JE, Sridhar SS, Zhang J, et al: Updated results from the enfortumab vedotin phase 1 (EV-101) study in patients with metastatic urothelial cancer (mUC). Journal of Clinical Oncology 36:4504-4504, 2018
- Siefker-Radtke AO, Necchi A, Park SH, et al: First results from the primary analysis population of the phase 2 study of erdafitinib (ERDA; JNJ-42756493) in patients (pts) with metastatic or unresectable urothelial carcinoma (mUC) and FGFR alterations (FGFRalt). J Clin Oncol 36, 2018