Introduction
Since the United States Food and Drug Administration (FDA) approval of mitoxantrone in 19961 and docetaxel in 20042 for the treatment of patients with metastatic castrate-resistant prostate cancer, we have witnessed the approval of numerous additional agents/combinations in this disease space:
- 2010: Sipuleucel-T3 and cabazitaxel4 (post-docetaxel)
- 2011: Abiraterone in the post-docetaxel setting5
- 2012: Abiraterone (pre-docetaxel)6 and enzalutamide (post-docetaxel)7
- 2013: Radium-2238
- 2014: Enzalutamide (pre-docetaxel)9
- 2017: Pembrolizumab (mismatch repair deficient [dMMR] or microsatellite instability-high [MSI-H])10
- 2020: Rucaparib (post-androgen receptor pathway inhibitor [ARPI] and taxane for BRCA-mutated)11 and olaparib (post-abiraterone or enzalutamide for patients with homologous recombination repair [HRR] alterations)12
- 2022: Lu-177 vipivotide tetraxetan (post-ARPI and taxane)13,14
- 2023: Olaparib + abiraterone (BRCA1/2-mutated)15 and talazoparib + enzalutamide (HRR-mutated)16
Despite these therapeutic advances with associated regulatory approvals, mCRPC patients have a poor prognosis, with an estimated median overall survival of approximately three years with currently approved first-line agents.2,6,9 In the real world, survival outcomes are even worse, with an estimated median survival of less than two years, likely due to patients in the real world often never receiving second- and third-line treatment regimens.17 Accordingly, there is an obvious need for novel therapeutic targets to improve survival outcomes in this patient cohort.
In this Center of Excellence article, we discuss emerging, novel therapeutic targets and/or applications of existing agents in the mCRPC disease space.
Androgen Receptor and Androgen Axis Targets
Androgen receptor (AR) gene amplification, commonly via tandem duplication, is the most frequent molecular alteration in mCRPC, occurring in about approximately 60%-70% of cases. Similar to gene amplification, structural rearrangements in the AR are also common in mCRPC, occurring in approximately 13 – 33% of patients prior to abiraterone or enzalutamide treatment and increasing in frequency to approximately 25 – 50% following treatment. These structural rearrangements can occur concomitantly with or independent of AR amplification and can give rise to diverse AR variant proteins with constitutive activity. Both AR gene amplification and structural rearrangements have been implicated in driving resistance to the currently approved 1st line mCRPC ARPIs, enzalutamide, and abiraterone.18,19
EPI-7386
EPI-7386 is an aniten, a class of compounds that inhibit AR activity by binding to the N-terminal domain, irrespective of resistance abrogation in the ligand binding domain that render ARPIs ineffective. Preclinical data supports disruption of AR regulated gene transcription even in the presence of resistance mechanisms including ligand-binding domain point mutations and truncated splice variants. Results of a first-in-human trial in heavily pre-treated mCRPC patients (NCT04221222) were recently presented at ASCO GU 2023. This was a phase 1, open-label, multicenter, dose escalation (Part 1a) and expansion (Part 1b) trial designed to evaluate the safety, pharmacokinetics, pharmacodynamics, and antitumor activity of EPI-7386 in mCRPC patients progressing on standard of care treatment, including ARPIs and chemotherapy. 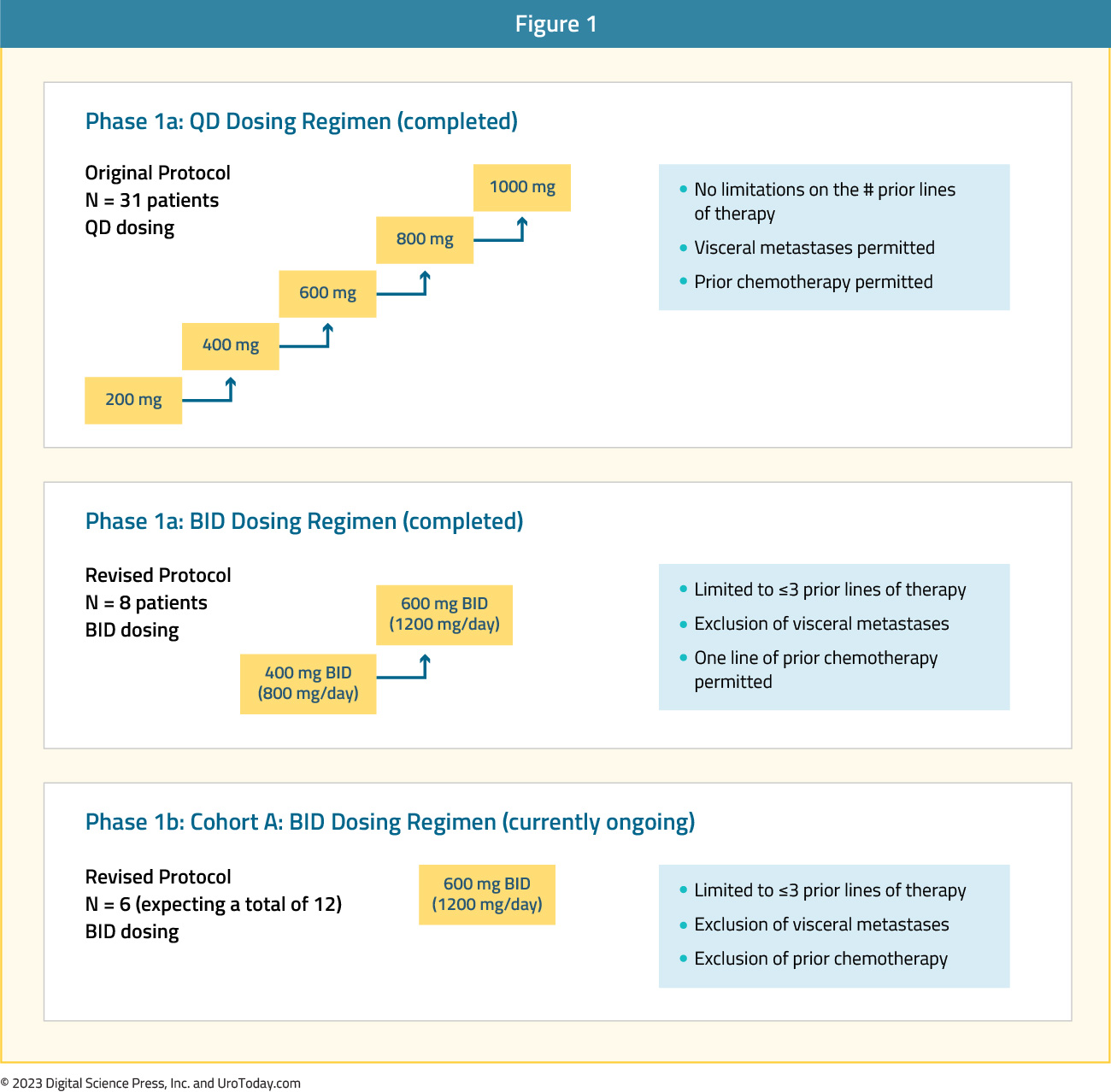
The phase 1a dose escalation portion of the trial included both daily (n=31) and twice daily dosing regimens (n=8). Patients in this trial were heavily pre-treated, with the median number of lines of prior therapy at 7 and 2 in the once and twice daily dosing cohorts, respectively (83% received abiraterone plus ≥1 other ARPI, and 58% had at least one line of prior chemotherapy).
No dose-limiting toxicities were observed; EPI-7386 was safe and well tolerated at all doses/schedules evaluated. All treatment-related adverse events considered grade 1 – 2 in severity (anemia, transaminitis, diarrhea, fatigue, hot flushes, nausea), except for one occurrence of grade 3 hypertension and anemia, both considered unlikely to be drug-related.
From an efficacy standpoint, evidence of antitumor activity (including significant and durable PSA responses and/or decreases in ctDNA, and/or radiographically documented tumor shrinkage) was observed in patients with <3 prior lines of mCRPC treatment, no visceral metastases, and no prior chemotherapy (9/31 patients). In the daily dosing, approximately 30% of patients remained on therapy for longer than 3 months, compared to 63% of patients in the twice daily dosing cohort.
Among the 25 patients who completed three cycles of treatment (12 weeks) without a PSA decrease, 10 (405) showed a reduction in PSA doubling time (PSADT), illustrated graphically below. Among nine patients with measurable nodal disease at baseline, seven experienced radiographic decrease in disease burden, even in the absence of PSA decline.20 The phase 1b dose expansion study is underway in a less heavily pre-treated population.
EPI-7386 + Enzalutamide
EPI-7386 has also been evaluated in combination with enzalutamide in ARPI-naïve, chemotherapy-exposed mCRPC patients. Preclinical RNAseq and CHIPseq data suggest that the combination of EPI-7386 plus enzalutamide results in a deep blockade of the AR pathway with greater antitumor activity compared to enzalutamide alone. In a phase 1/2 multicenter, open-label clinical trial (NCT05075577), escalating doses of EPI-7386 were tested in combination with a fixed dose of enzalutamide (120 mg daily; phase 1 portion), with primary and secondary endpoint of safety and pharmacokinetics with the goal of determining the recommended phase 2 combination dose. This is particularly relevant given that EPI-7386 is a known inhibitor of CYP2C8, a metabolizer of enzalutamide, thus potentially increasing patient exposure to enzalutamide. Conversely, enzalutamide is a known inducer of CYP enzymes that participate in the metabolism of EPI-7386, thereby potentially decreasing patient exposure to the novel drug.
Results of the first seven treated patients, including three from Cohort 1 (EPI-7386 600 mg QD + enzalutamide 120 mg QD) and four from Cohort 2 (EPI-7386 800 mg QD + enzalutamide 120 mg QD), were presented at ASCO GU 2023. No dose-limiting toxicities were observed, and the safety profile was consistent with those of second-generation antiandrogen, including grade 1-2 fatigue and hot flashes.
From a pharmacokinetics standpoint, enzalutamide decreased EPI-7386 exposure, likely via increased metabolism, whereas enzalutamide pharmacokinetics were not altered by EPI-7386 dosing. Efficacy data was available for 6/7 patients. Five (83%) patients achieved a PSA decline of at least 90% (PSA90), with four (67%) attaining a PSA of <0.2 ng/mL (all within 90 days). All six of these patients demonstrated stable imaging.21
Relacorilant
Glucocorticoid receptor expression increases with AR inhibition and its blockade inhibits CRPC growth in preclinical models when combined with AR blockade. A phase 1 study (NCT03674814) evaluating the combination of relacorilant, a novel glucocorticoid receptor antagonist, and enzalutamide in mCRPC patients was recently presented at ASCO 2023.
This phase 1 study employed a 6+3 design to assess the safety and pharmacokinetics of this combination. The starting doses of relacorilant and enzalutamide were 100 mg and 120 mg daily, respectively, based on anticipated drug-drug interaction. Escalating doses of relacorilant were combined with enzalutamide 120 mg using pre-specified dose escalation rules.
Thirty-five patients were enrolled with a median age of 74 years and a baseline PSA of 89.5 ng/ml. All patients had received a prior ARPI, and 69% had received prior docetaxel. The maximum tolerated dose was determined to be relacorilant 150 mg + enzalutamide 120 mg. The most common grade ≥3 treatment-related adverse events were fatigue (11%), anemia (9%), abdominal pain (6%), and pericardial effusion (6%). From an efficacy standpoint, the median progression-free survival was 2.5 months among the 21 evaluable patients.22
The best responses per RECIST 1.1 were:
- Not evaluable: 40%
- Stable disease: 34%
- Progressive disease: 26%
- Complete or partial response: 0%
ODM-208
ODM-208 is a novel, oral, non-steroidal, and selective inhibitor of CYP11A1, the first and rate-limiting enzyme of steroid biosynthesis. Through inhibition of CYP11A1, ODM-208 suppresses the production of all steroid hormones and their precursors that may activate the AR signaling pathway. This mechanism of action is particularly relevant for patients with activating somatic point mutations in the AR ligand-binding domain. Such mutations are a known mechanism of resistance to hormone-based therapies in mCRPC.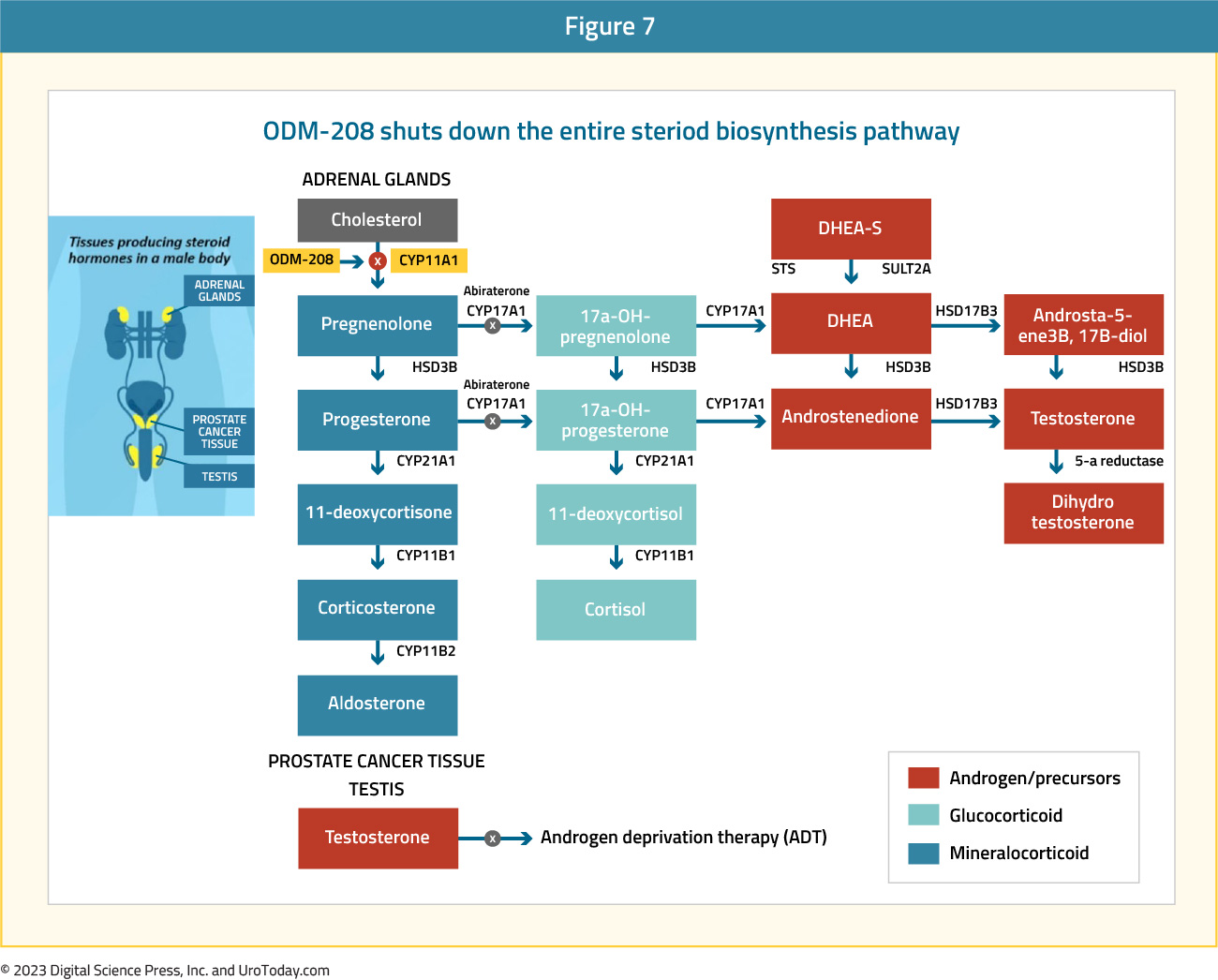
The CYPIDES trial utilized a phase I dose-finding 3+3 design among men with progressive mCRPC who had previously received ≥1 ARPI and ≥1 taxane. Patients received ODM-208 up to 150 mg/day, with glucocorticoid and mineralocorticoid replacement therapy, plus ADT.
The results of this phase 1 trial were presented at ASCO GU 2022. There were 44 patients included, of whom 55% had previously received both abiraterone and enzalutamide, and all had received ≥1 taxane-based chemotherapy. With the accrual of 41 patients, dose-finding was completed with doses ranging from 10 to 150 mg/day. Although tolerated by most patients, the main safety finding was adrenal insufficiency. Overall, 15/41 (37%) patients experienced Grade 3 adrenal insufficiency, requiring short-term high dose glucocorticoid treatment. Given that ODM-208 was more potent than initially predicted, the dose was de-escalated.
In terms of biochemical correlates of treatment, serum testosterone was undetectable after 4 weeks of start of ODM-208 in nearly all patients, as were other metabolites including serum DHEA sulfate, androstenedione, 11β-hydroxyandrostenedione, 11-ketotestosterone, and pregnenolone. From an efficacy standpoint, 32% of evaluable patients achieved a PSA decline of ≥50%. This rate was higher (68%) in patients with activating somatic point mutations in the AR ligand-binding domain than in those men without (8%).23

Bavdegalutamide (ARV-110)
Oral bavdegalutamide (ARV-110) is a PROteolysis Targeting Chimera (PROTAC) AR degrader that has activity against clinically relevant mutations via creation of a trimer complex with AR and the cereblon E3 ubiquitin ligase to directly trigger ubiquitination and subsequent degradation of AR by the proteasome: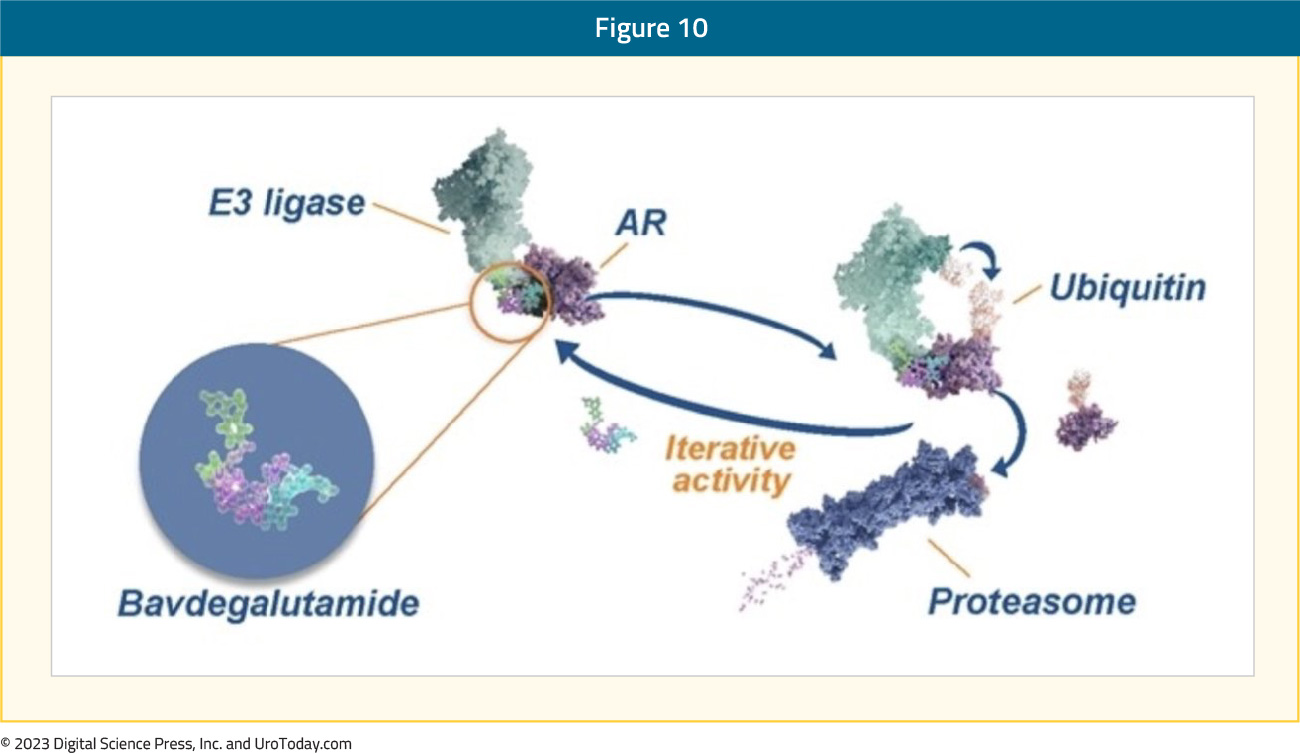
Additionally, bavdegalutamide has shown clinical activity in previously treated mCRPC patients in a phase 1/2 study (NCT03888612), with enhanced activity seen in patients with the AR ligand binding domain mutations T878 and/or H875.
In a phase 1/2 study of bavdegalutamide, patients with mCRPC and ≥2 prior therapies (including an ARPI) received bavdegalutamide 35–700 mg once daily or 210–420 mg twice daily (phase 1). In the phase 2 portion, patients with mCRPC and 1–2 ARPIs +/- prior chemotherapy were assigned to a biomarker-defined subgroup and received bavdegalutamide 420 mg once daily. In exploratory analyses across the phase 1/2 population treated with 420 mg bavdegalutamide once daily, efficacy was assessed in patients with AR 878/875 mutations without an AR L702 mutation (AR 878/875 patients) and in those with any AR ligand binding domain missense mutation except AR L702H alone (AR ligand binding domain patients).
Presented at ESMO 2023, 153 patients across the phase 1/2 study received bavdegalutamide 420 mg daily, including 45 patients with AR ligand binding domain mutations excluding L702H alone, 26 of whom had AR 878/875 without L702H. The median radiographic progression-free survival per PCWG3 was 8.2 months in AR ligand binding domain patients (n=45) and 11.1 months in the AR 878/875 subgroup (n=26):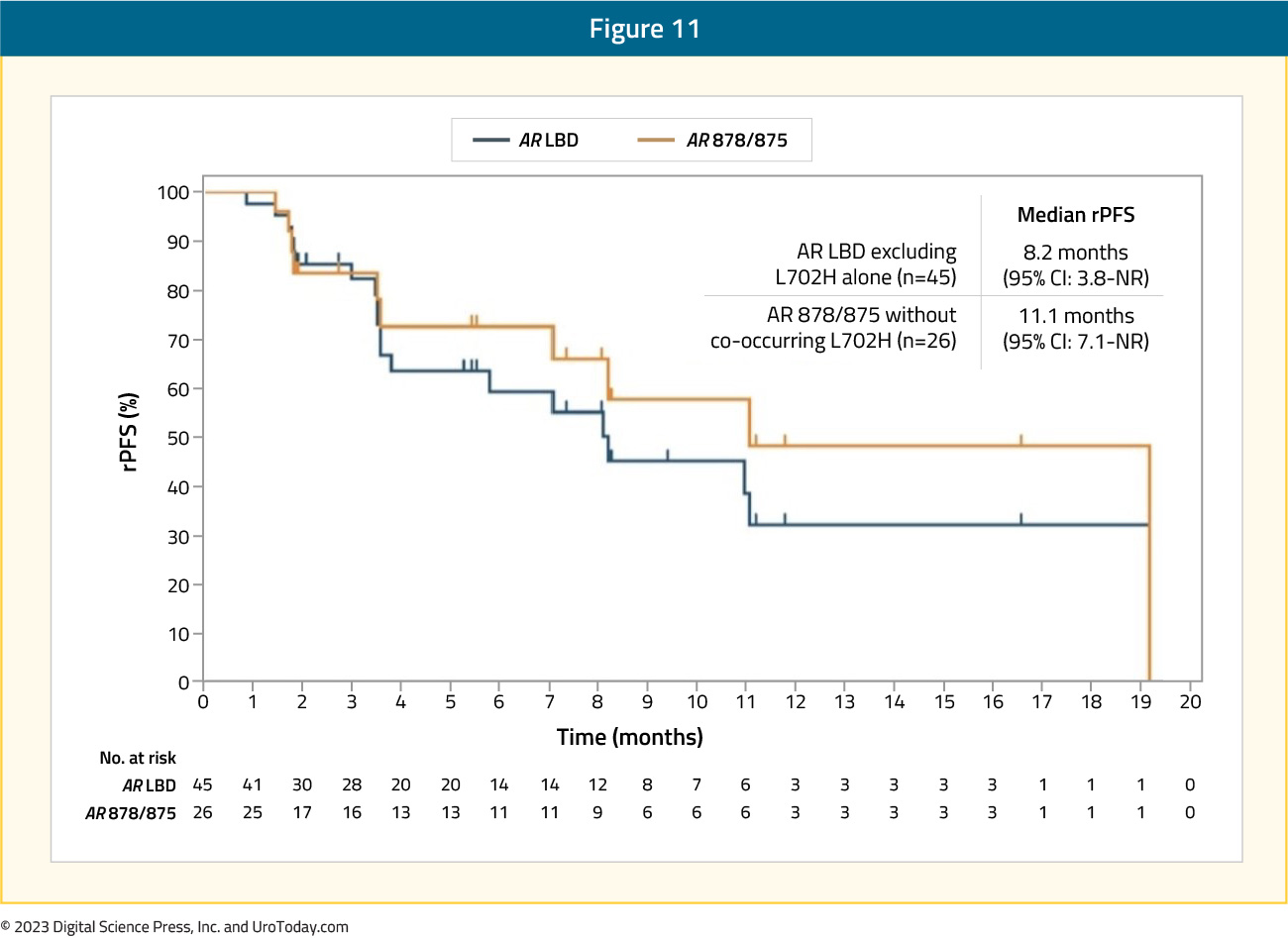
PSA declines of ≥50% and ≥30%, respectively, after ≥1 month of PSA follow-up were seen in 36% and 50% of AR ligand binding domain patients (n=44) and 54% and 65% of patients in the AR 878/875 subgroup (n=26):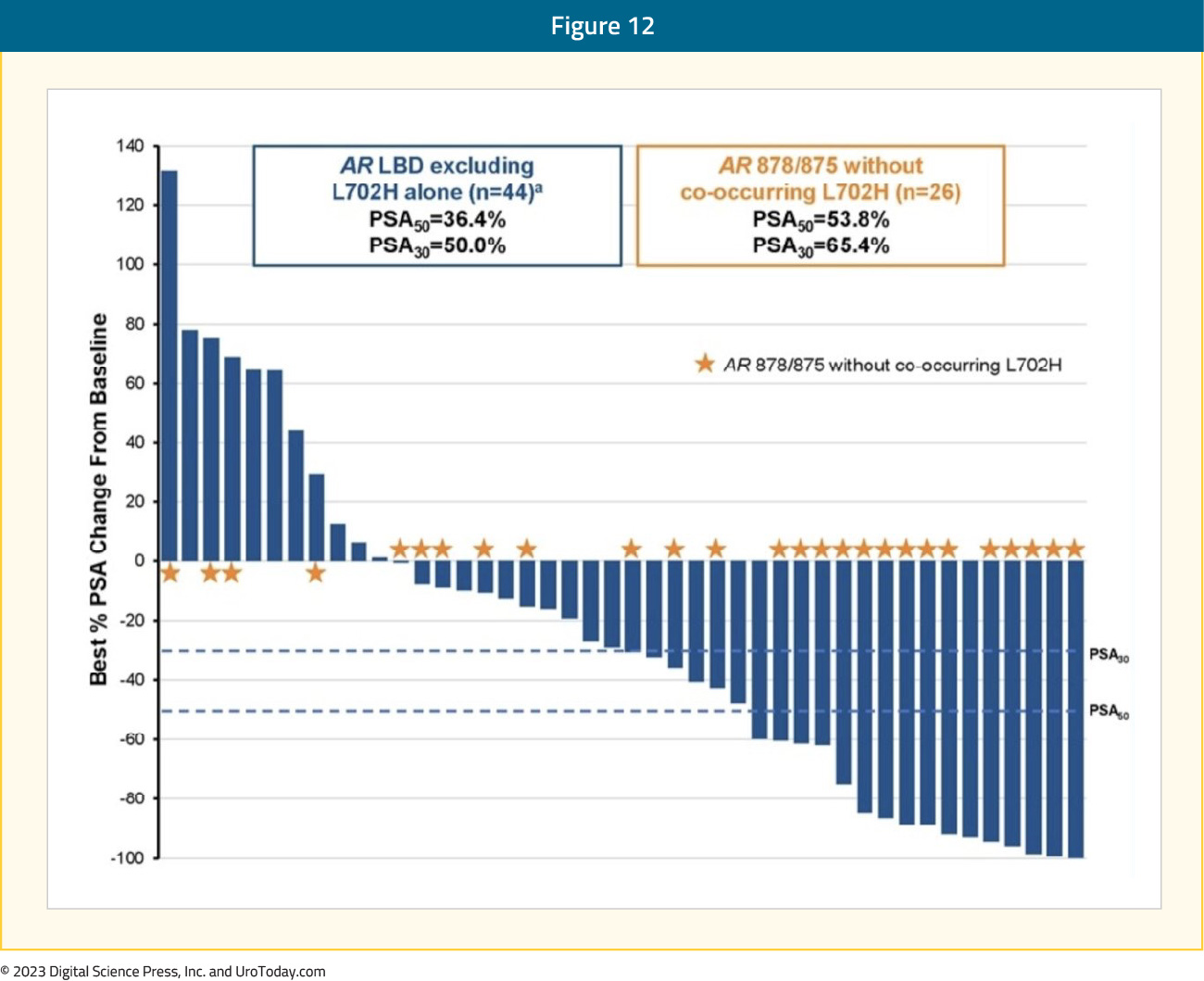
In response-evaluable patients with measurable disease at baseline, overall response rate was 10% in AR ligand binding domain (n=20) patients, and 9% in AR 878/875 patients (n=11); 55% and 54.5%, respectively, had stable disease. Across the phase 1/2 study, 147 (96%) of 153 patients had a treatment emergent adverse event, of which 47 (31%) had a grade 3/4 event. Overall,17 (11%) of patients had a treatment emergent adverse event that led to a dose reduction and 19 (12%) led to discontinuation. The most common treatment emergent adverse events were nausea, fatigue, and vomiting.24
Abemaciclib
AR signaling promotes translation of D-type cyclins resulting in cyclin-dependent kinase 4 and 6 (CDK4 and 6) activation and cell cycle progression. Abemaciclib is an oral selective inhibitor of CDK4 and 6 that is FDA-approved in combination with endocrine therapy or as monotherapy to treat HR+, HER2- metastatic breast cancer patients. Preclinical studies using prostate cancer cell lines and xenograft models have shown that abemaciclib induces cell cycle arrest and tumor growth inhibition. It has been hypothesized that the addition of abemaciclib to AR-targeted therapy may be an effective treatment for mCRPC patients.
CYCLONE 2 (NCT03706365) is a phase 2/3, randomized, double-blind, multicenter, placebo-controlled study assessing the safety and efficacy of abemaciclib in combination with abiraterone acetate plus prednisone in mCRPC patients. CYCLONE 2 is an adaptive study that is designed in three parts:
- Part 1 is a 30-patient safety lead-in to determine the recommended phase 2 dose (150 mg or 200 mg, twice daily) of abemaciclib in combination with abiraterone acetate (1000 mg, once daily) + prednisone (5 mg, twice daily)
- In part 2, 150 patients are randomized 1:1 to abiraterone acetate plus prednisone with abemaciclib at the recommended phase 2 dose or placebo
- The study expands to enroll an additional 170 patients in Part 3 if prespecified expansion criteria are met at a planned adaptive interim analysis performed by an independent data monitoring committee

Systemic anti-cancer therapy for mCRPC and prior novel hormonal agents are exclusionary. Prior docetaxel in the hormone-sensitive setting is permissible. The primary objective is radiographic progression free survival, with secondary objectives of safety, objective response rate, duration of response, time to symptomatic and PSA progression, overall survival, and pharmacokinetics. Currently, enrollment in Parts 1 and 2 is completed. Based on the recommendation from the independent data monitoring committee, Part 3 was opened in June 2021 and enrolled patients from 112 sites across 12 countries.24
Chimeric Antigen Receptor-Engineered T (CAR-T) Cell Therapy
Chimeric antigen receptor T (CAR-T) cell therapy is being successfully used to treat hematological malignancies, such as lymphomas and leukemias. CAR-T technology is currently being evaluated in prostate cancer expressing PSMA and/or prostate stem cell antigen (PSCA), which is highly expressed on the surface membrane in mCRPC but has limited expression on normal tissues, making it an ideal target for therapy. PSCA-targeted 4-1BB-co-stimulated CAR T cell therapy has recently been investigated in a phase 1, first-in-human study.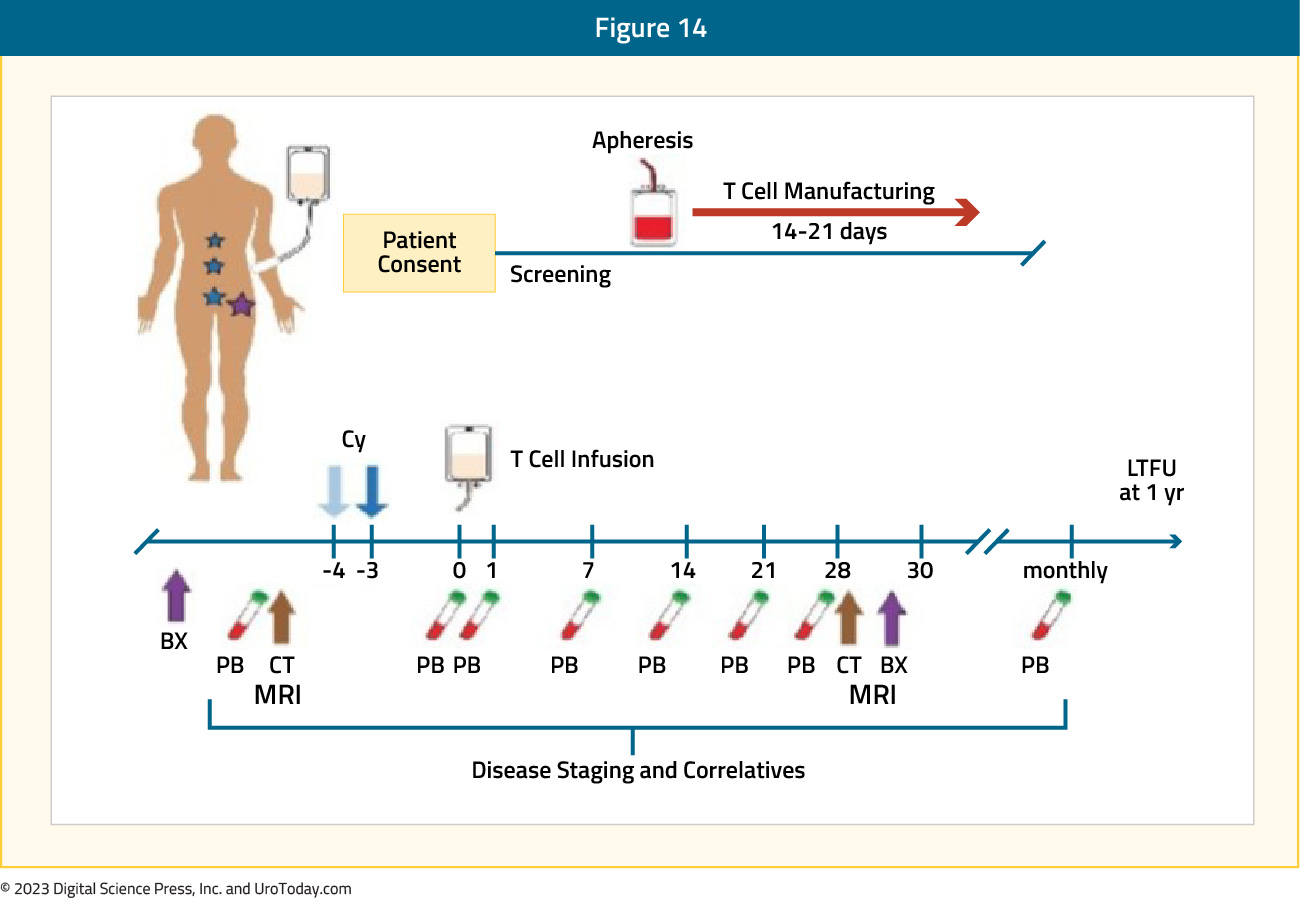
This trial included 14 mCRPC patients with disease progression following ≥1 ARPI (no limit on prior chemotherapy). The median baseline PSA was 88 ng/ml. Of note, 79% of screened patients had PSCA expression >2+ in >80% of cancer cells. The major dose-limiting toxicity was grade 3 cystitis, encountered in 2/6 patients receiving CAR-T therapy + lymphodepletion chemotherapy (cyclophosphamide and fludarabine). The protocol was amended to reduce the chemotherapy dose, and no further dose-limiting toxicities were noted.
From an efficacy standpoint, the best objective response per RECIST was stable disease in 7/14 patients, with 6 having progressive disease and none having a partial or complete response (1 non-evaluable for response). A PSA decrease by day 28 was observed in 4/15 patients only.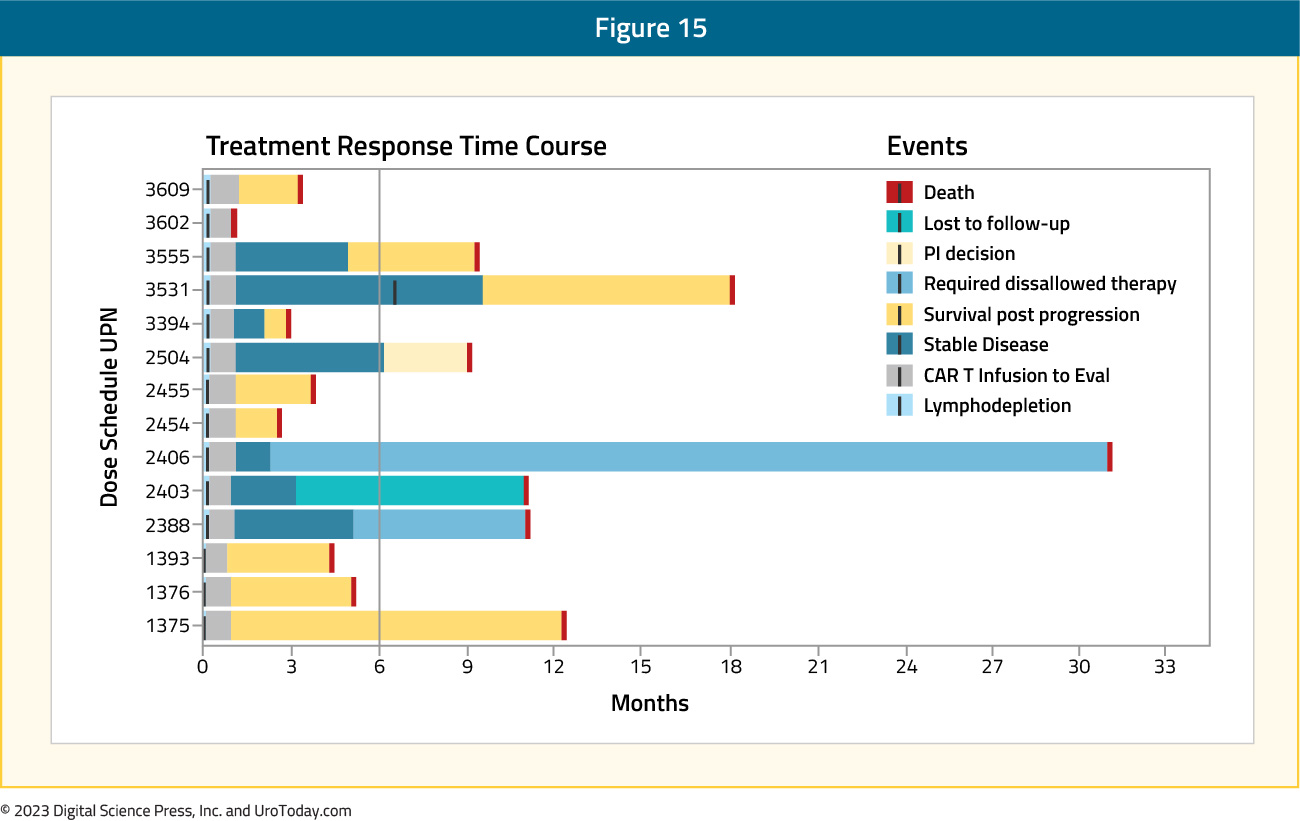
Prostate-Specific Membrane Antigen Targets ARX517
Initial PSMA-targeted antibody drug conjugates demonstrated early clinical efficacy, but drug development was discontinued due to intolerable toxicities, resulting from premature release and off-target delivery of the cytotoxic payload. ARX517 is a novel antibody-drug conjugate composed of a fully humanized anti-PSMA monoclonal antibody conjugated to amberstatin-269, a potent tubulin inhibitor. ARX517 was designed to reduce off-target antibody-drug conjugate instability-related toxicity issues observed in earlier anti-PSMA antibody-drug conjugates by using site-specific synthetic amino acids and stable oxime conjugation chemistry to minimize premature payload release in the human circulation.
APEX-01 is a first-in-human phase 1/2 study of ARX517 that used an i3+3 dose escalation design, with eligible patients having received ≥ 2 FDA-approved treatments for mCRPC with evidence of progression by Prostate Cancer Working Group criteria:
The initial safety and efficacy results were recently presented at ESMO 2023. This analysis included 65 patients who received ARX517 every 3 weeks at escalating doses. Patients had a median of four prior lines of therapy, 100% had received an ARPI, 66% received a taxane, and 17% received PSMA-targeted radionuclide therapy. Grade 1-2 treatment-related adverse events included dry mouth (28%), fatigue (20%), and diarrhea (15%). There were 6 (9%) patients that had grade 3 adverse events. No dose limiting toxicities, treatment-related serious adverse events, or ≥ grade 4 adverse events were reported.
Overall, 52% (12/23) of patients experienced ≥50% PSA reduction at putative therapeutic doses (≥ 2.0 mg/kg):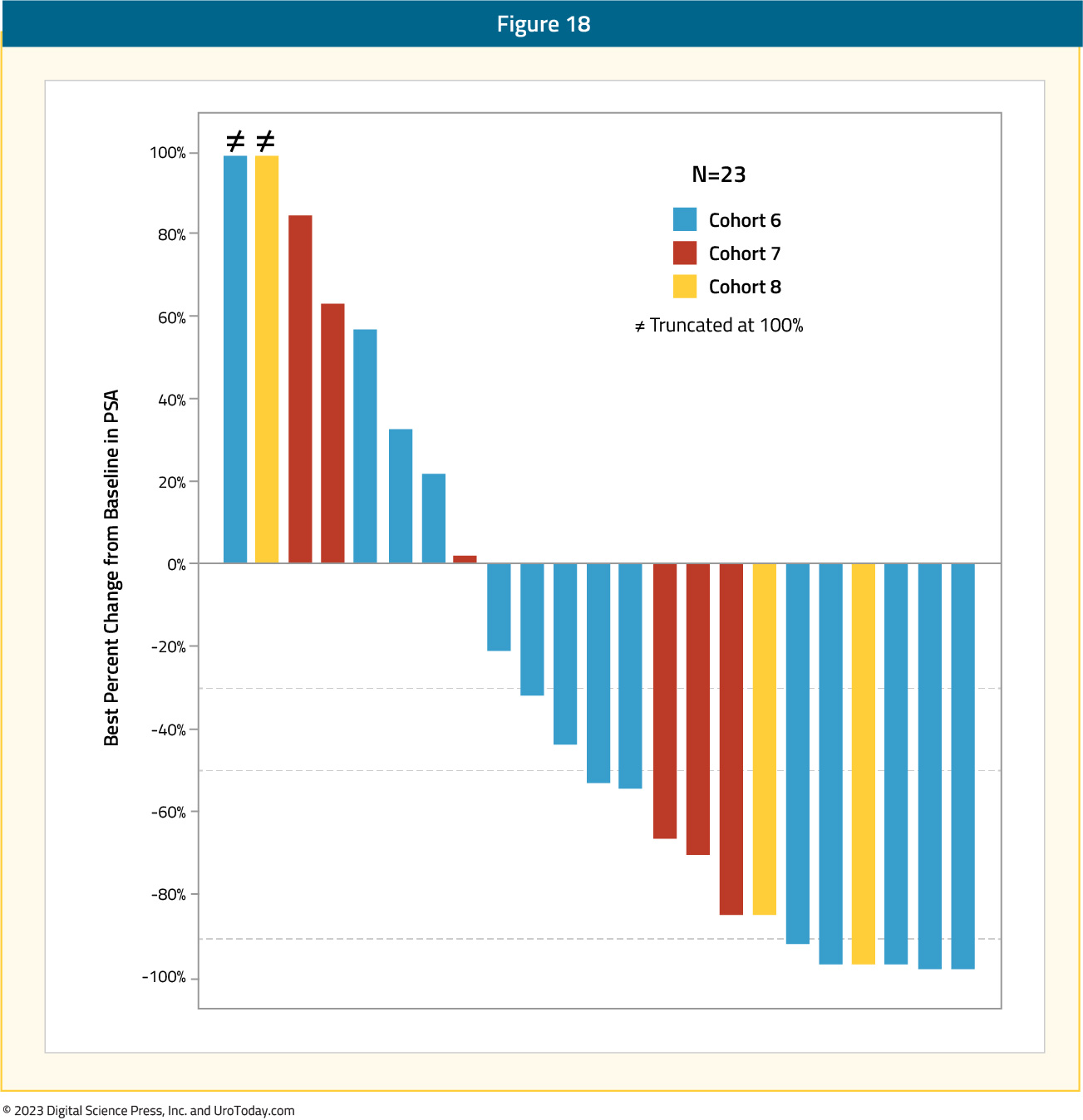
RECIST v1.1 target lesion reduction was observed in 56% (5/9) of patients in cohorts 4-8: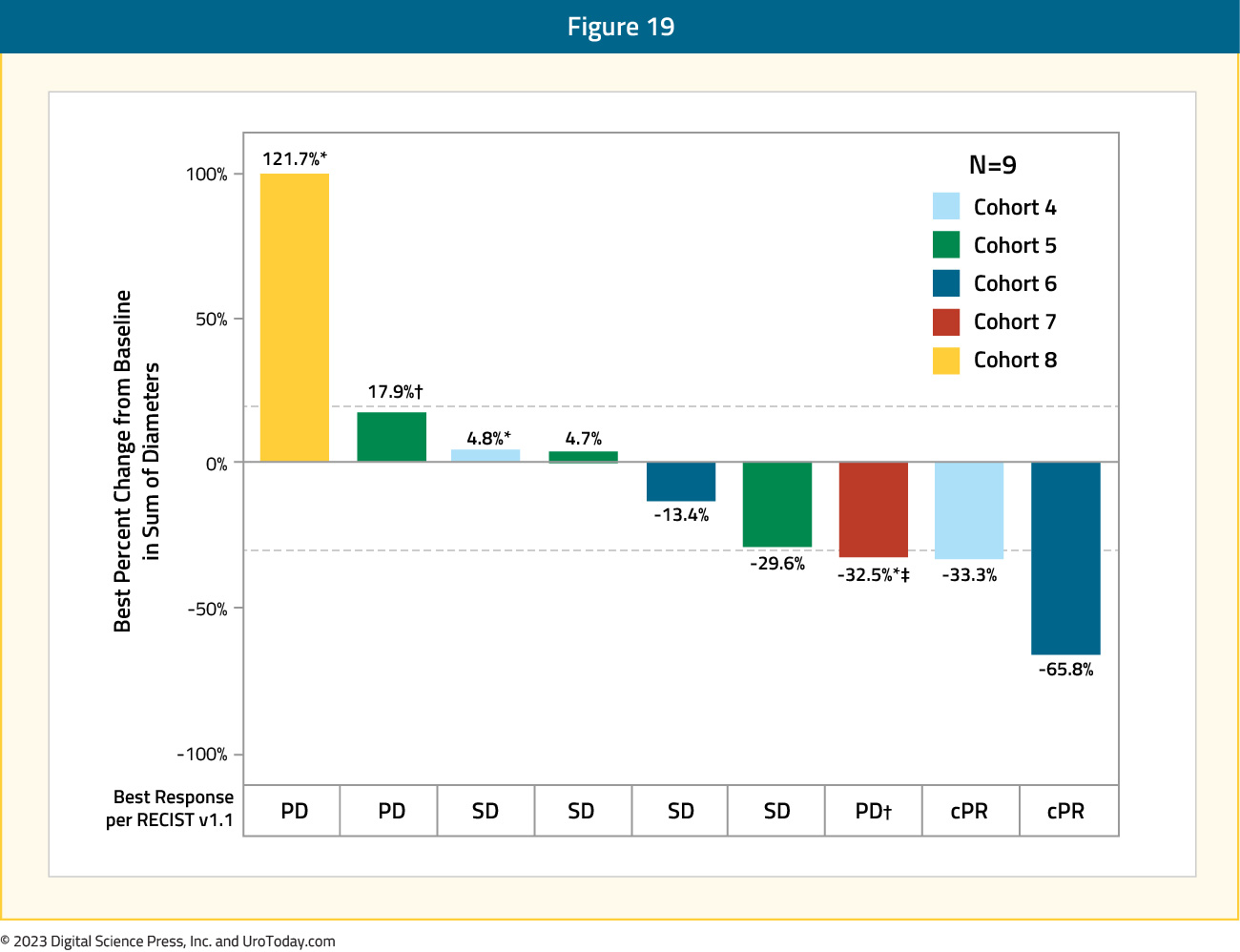
Based on these results, the investigators of APEX-01 concluded that ARX517 monotherapy achieved a favorable safety profile and demonstrated early efficacy, with deep PSA and confirmed RECIST v1.1 tumor responses in men with mCRPC who progressed on multiple FDA-approved treatments.
Lu-PSMA
177Lu-PSMA-617 is a targeted radioligand therapy for PSMA-positive mCRPC with the following mechanism of action: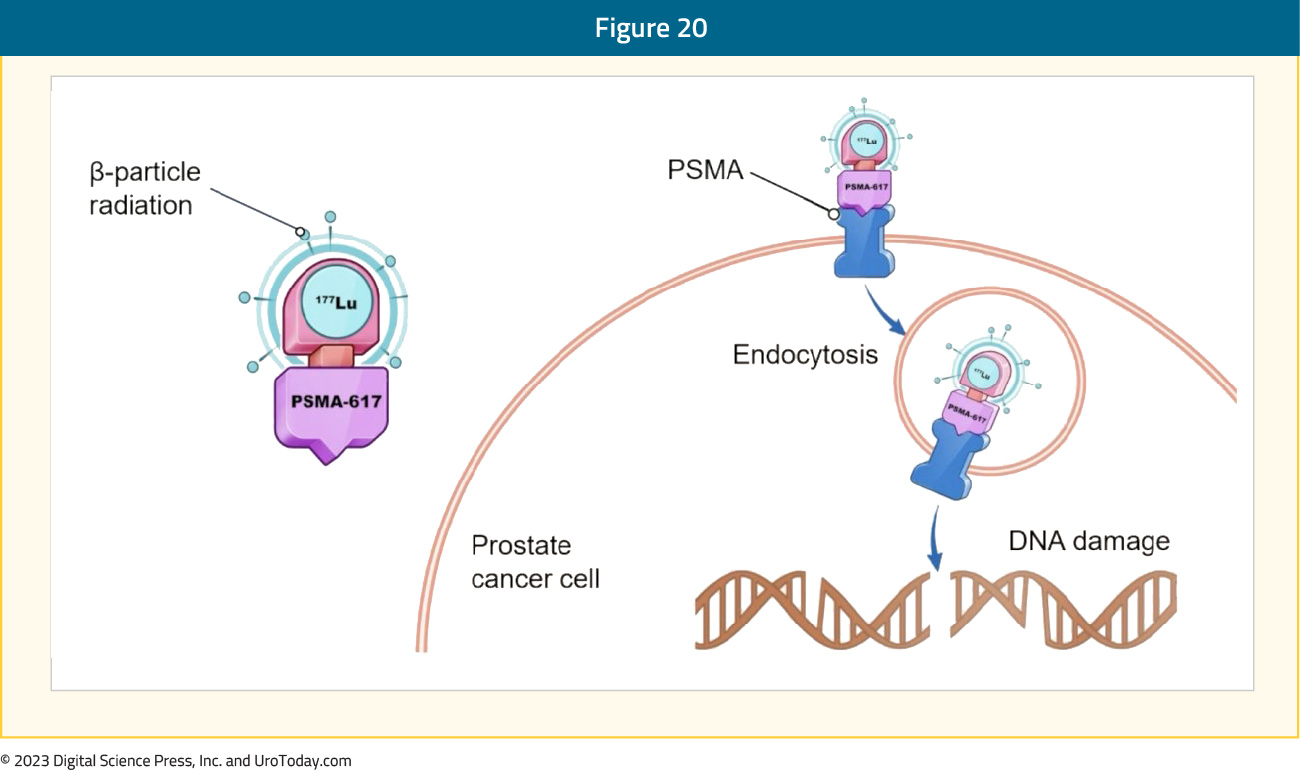
Following the approval of Lu-PSMA in the post-ARPI and docetaxel mCRPC setting, there has been increased interest in moving this therapy further up the mCRPC treatment landscape. The PSMAfore trial subsequently examined 177Lu-PSMA-617 in taxane-naive patients and was presented at the ESMO 2023 annual congress.
PSMAfore (NCT04689828) is a phase III, open-label, multi-center, randomized study comparing 177Lu-PSMA-617 to an ARPI change (abiraterone or enzalutamide) in taxane-naïve mCRPC patients. All eligible patients were candidates for an ARPI change following progression on one ARPI and had ≥1 PSMA positive lesion(s) and no exclusionary PSMA negative lesions by 68Ga-PSMA-11 PET/CT. Candidates for PARP inhibition and patients with prior systemic radiotherapy (<6 months ago), immunotherapy (except sipuleucel-T), or chemotherapy (except [neo]adjuvant >12 months ago) were ineligible. Randomization was 1:1 to open-label 177Lu-PSMA-617 (7.4 GBq every 6 weeks for 6 cycles) or an ARPI change (abiraterone or enzalutamide). Importantly, patients randomized to ARPI could crossover to 177Lu-PSMA-617 following centrally reviewed radiographic progression. The trial design for PSMAfore is as follows:
Overall, there were 468 patients randomized. The median patient age was 71 – 72 years and median PSA was 14.9 – 18.4 ng/ml. Notably, 84.2% of patients that had an rPFS event (primary endpoint) in the ARPI change arm subsequently went on to receive crossover 177Lu-PSMA-617. At the primary analysis (median follow-up, 7.3 months; n=467), the primary endpoint of rPFS was met (HR:0.41, 95% CI: 0.29 to 0.56).
Among men with measurable disease, the objective response rate was 51% versus 15% in favor of the 177Lu-PSMA-617 arm (complete response: 21% versus 3%). The median duration of response was 13.6 months for 177Lu-PSMA-617 versus 10.1 months for ARPI change. To date, overall survival data remains immature (45.1% of death events have occurred). In the pre-specified cross-over adjusted analysis, there was a trend towards an overall survival benefit in the 177Lu-PSMA-617 arm (HR: 0.80. 95% CI: 0.48 to 1.33). In the unadjusted, intention to treat analysis, the hazard ratio was 1.16 (95% CI: 0.83 to 1.64).
For 177Lu-PSMA-617 versus ARPI change, the incidence of grade ≥3 adverse events was 34% (most commonly anemia and dry mouth) versus 43%, serious adverse events 20% versus 28%, and adverse events leading to discontinuation 5.7% vs 5.2%, respectively. A summary of the most common adverse events is as follows:
The phase III SPLASH trial (NCT04647526) is similarly evaluating 177Lu-PNT2002 in the 2nd line, post-ARPI mCRPC treatment setting with final results expected to be available in early 2024.
Lu-PSMA Combinations
177Lu-PSMA-617 + Olaparib
Beta radiation delivered to PSMA-expressing tumors via 177Lu-PSMA-617 results in single strand DNA breaks, which are typically repaired by Poly (ADP-ribose) polymerase
(PARP)-dependent pathways. Blocking PARP could result in the conversion of DNA single strand breaks to lethal double strand breaks via replication fork collapse. In the LuPARP trial, it was hypothesized that the PARP inhibitor olaparib could promote radiosensitization to 177Lu-PSMA-617, enhancing resultant DNA damage and improving efficacy.
This study followed a 3+3 dose escalation design, with the trial schema illustrated below:
This trial included 48 patients with mCRPC, all of whom had received a prior ARPI and docetaxel. All patients underwent a 68Ga-PSMA-11 plus an FDG-PET/CT with the following inclusion criteria:
- PSMA SUVmax >15 at any site
- SUVmax >10 at other sites
- No FDG discordance
No dose limiting toxicities were observed, and the recommended phase 2 dose was established at 7.4 Gb of 177Lu-PSMA-617 in conjunction with olaparib 300 mg twice daily on days -4 to 18 of each 6-weekly cycle. Three patients (9%) required a dose delay due to hematologic toxicity, and dose reductions were required in four patients (hematologic toxicity: 3, xerostomia: 1). There were no grade 4 adverse events.
Across all evaluable cohorts, the PSA50 and PSA90 response rates were 66% and 44%, respectively. The objective response rate by RECIST v1.1 criteria was 78%. The PSA50 responses from this trial were identical to those from TheraP (66%) and higher than those in VISION (46%). The PSA90 response of 44% in LuPARP was slightly higher than that in TheraP (38%).26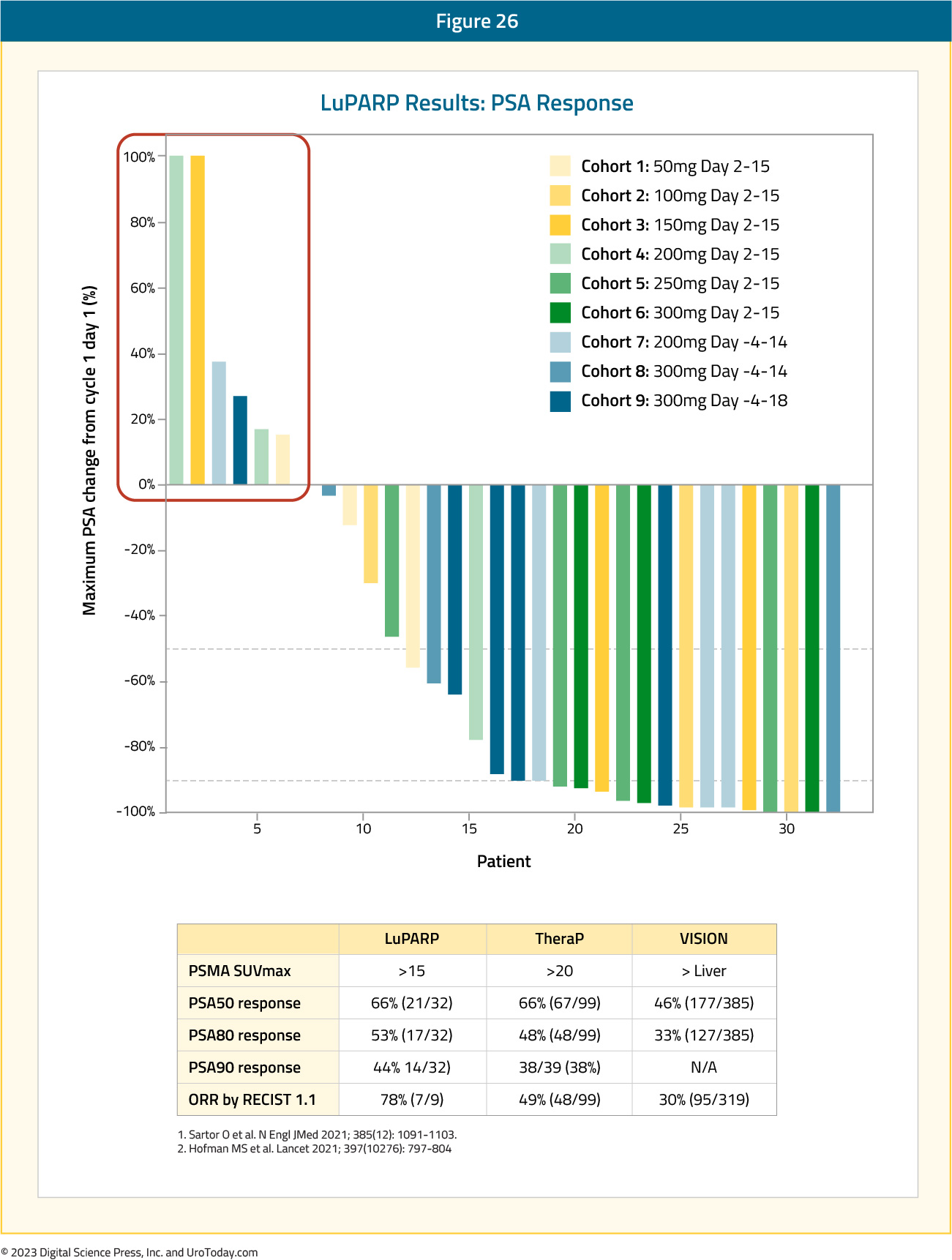
Preclinical and clinical data suggest synergy between Lu-PSMA and ARPIs in mCRPC. Specifically, the clonal adjustment theory suggests that enzalutamide increases androgen resistance, allowing LuPSMA to have enhanced activity. ENZA-p (ANZUP 1901) is a phase 2 trial of enzalutamide plus 177Lu-PSMA-617 in poor-risk mCRPC patients who had no prior treatment with chemotherapy or ARPIs for castrate-resistant disease (prior abiraterone and/or docetaxel for hormone-sensitive disease was permitted), 68Ga-PSMA-positive disease on PET, and ≥2risk factors associated with early progression on enzalutamide:
- LDH ≥ upper limit of normal
- ALP ≥ upper limit of normal
- Albumin <35 g/L
- De novo metastatic disease at diagnosis
- <3 years since initial diagnosis
- >5 bone metastases
- Visceral metastases
- PSA doubling time <84 days
- Prior abiraterone
The trial design for Enza-P is as follows: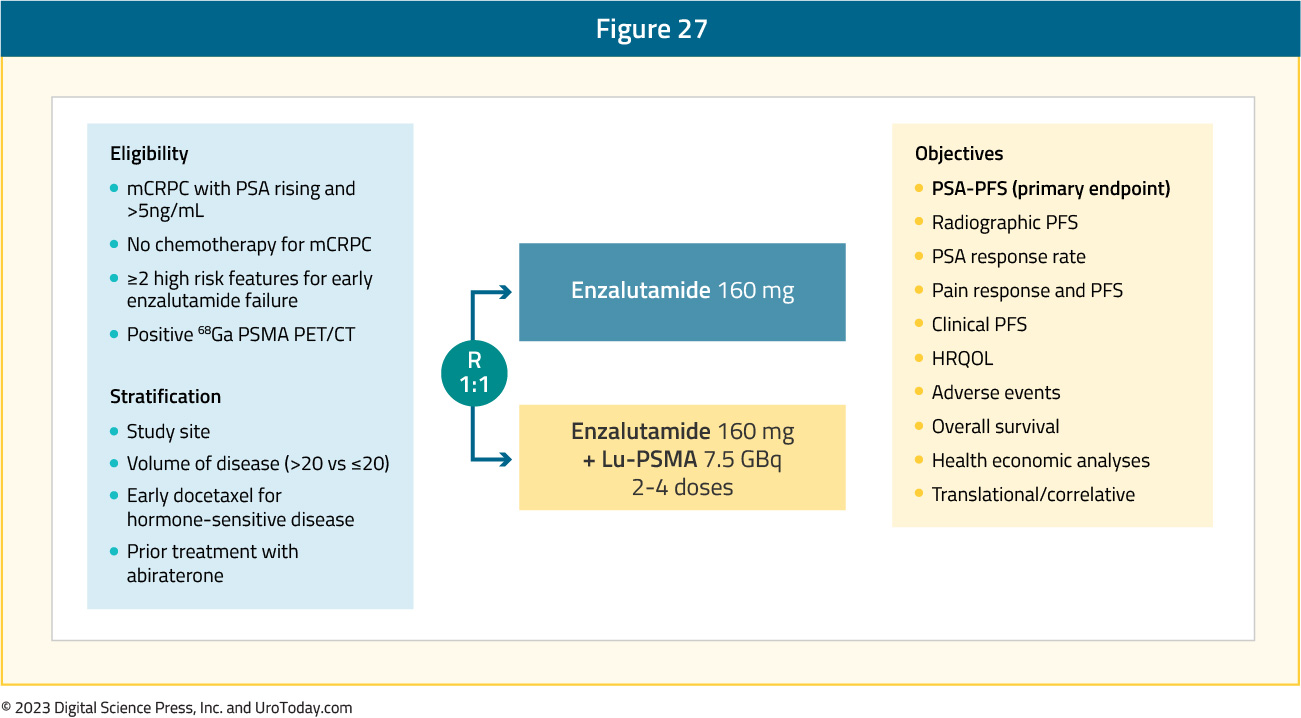
This trial included 162 patients, of whom 83 and 79 were randomized to the enzalutamide + Lu-PSMA and enzalutamide arms, respectively. The median age was 71 years and prior docetaxel and abiraterone were each used in 54% and 13% of men, respectively.
The combination of Lu-PSMA + enzalutamide significantly prolonged PSA-PFS, the primary study endpoint, by over 5 months (median: 13 versus 7.8 months; HR: 0.43, 95% CI: 0.29 – 0.63, p<0.001).
The combination of Lu-PSMA + enzalutamide was also associated with significant improvements in:
- Radiographic PFS (HR: 0.67, 95% CI: 0.44 to 1.01)
- PSA50 (93% versus 68%) and PSA90 (78% versus 37%) responses
Symptomatic adverse events were reported in 33%of patients assigned enzalutamide + LuPSMA versus 35% of those in the enzalutamide-alone arm. As follows is a tornado plot summarizing the adverse events of interest: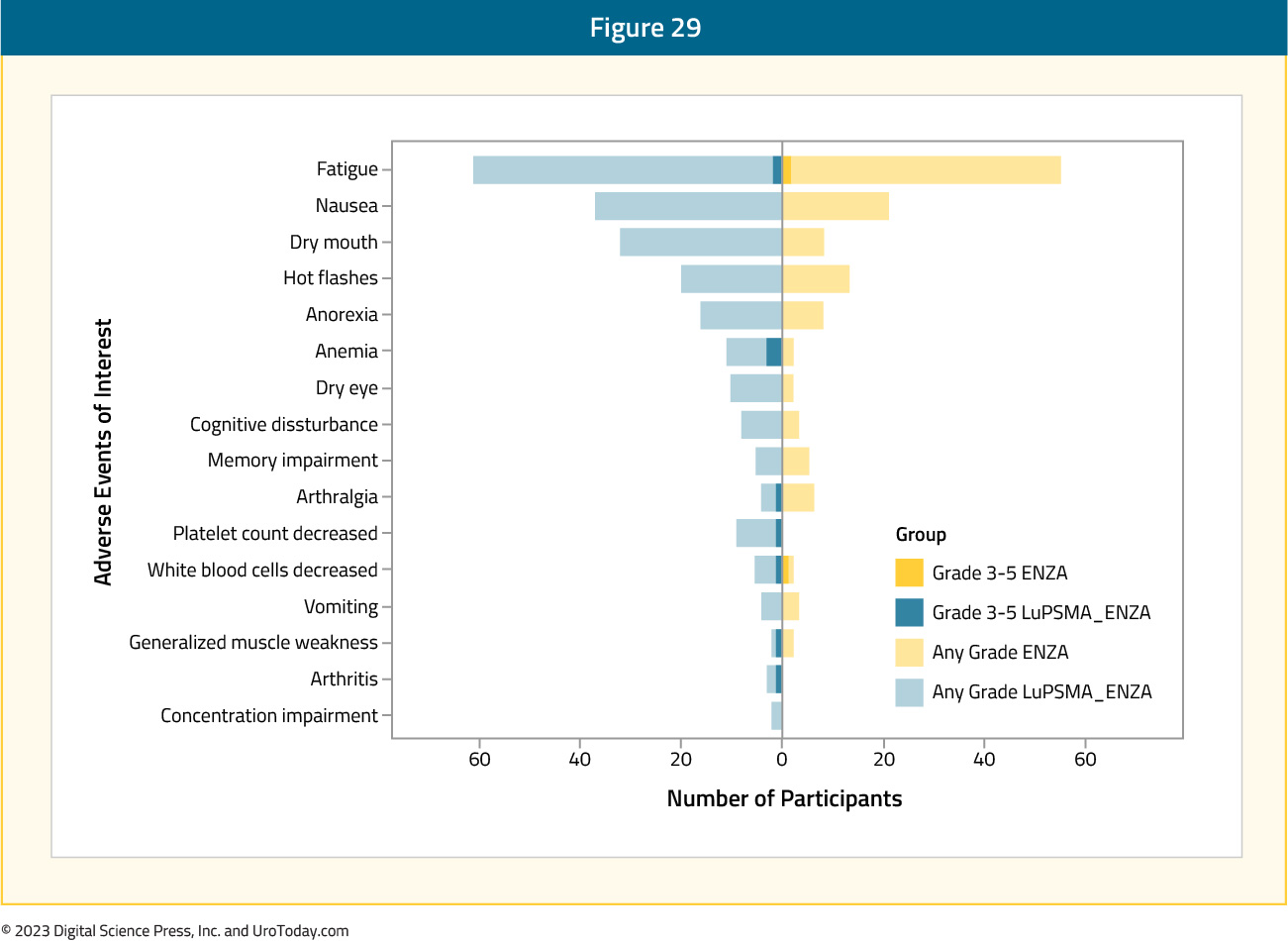
The rationale for combining 177Lu-PSMA-617 and cabazitaxel is that 177Lu-PSMA-617 may not effectively target micrometastatic disease due to the longer path length of beta emitters, which could be addressed with systemic cabazitaxel. Conversely, cabazitaxel has radiosensitizing properties that may enhance the cytotoxic effects of 77Lu-PSMA-617, while also treating any PSMA-negative disease.
The ongoing LuCAB single arm phase I/II trial is evaluating the combination of 177Lu-PSMA-617 and cabazitaxel in mCRPC patients with disease progression following prior ARPI and docetaxel exposure and evidence of PSMA-positive disease on PSMA-PET/CT (SUVmax ≥15). The target sample size is 32 – 38 patients. Up to 6 doses of 177Lu-PSMA-617 (7.4 GBq) will be administered intravenously every 6 weeks. Cabazitaxel will be given concurrently (dose range 12.5mg/m2 - 20mg/m2), on Day 2 and Day 23 of each 6-week cycle:
The primary objective is to establish the maximum tolerated dose of cabazitaxel and [177Lu-PSMA-617. Secondary objectives include:
- Measuring the frequency and severity of adverse events
- Assessment of efficacy through PSA 50% response rate
- Radiographic and PSA progression-free survival
- Overall survival
- Objective tumor response rate
- Evaluation of pain and health-related quality of life over the first 12 months
As of January 2023, five patients have been enrolled in the study, with preliminary results expected in the upcoming few years.27
Ac-225-PSMA-617
Targeted alpha emitters, such as Ac-225 PSMA radioligand therapy, have the potential to induce cell damage even following radioresistance to beta emitters such as 177Lu-PSMA-617, owing to their higher linear energy transfer and different microdosimetry in tumor tissue. By efficiently inducing increased DNA double-strand breaks, targeted alpha therapy has been more effective than targeted therapy with beta emitters in preclinical studies.28
225-Ac PSMA-617 is an alpha emitter with a t½ of 10 days. This agent has an associated higher energy deposition and shorter path length compared to the beta emitter Lu-PSMA, with the potential to:
- Overcome radiation resistance in Lu-PSMA resistant patients
- Improve responses with low(er) PSMA expression
- Better treat micrometastatic disease

In a meta-analysis of six retrospective studies, including a total of 201 patients,29 treatment with 225-Ac PSMA-617, commonly following prior Lu-PSMA treatment failure, was associated with the following pooled outcomes:
- PSA50 response: 66% (95% CI: 60 – 73%)
- Overall survival: 12.5 months (95% CI: 6.2 – 18.8 months)
- Progression-free survival: 9.1 months (95% CI: 2.6 – 15.7 months)
The most common adverse events were:
- Xerostomia: 77.1% (G3 in 3%)
- Anemia: 30.3% (G3 in 7.5%)
- Thrombocytopenia:14.9% (G3 5.5%)
- Nephrotoxicity (G3 3%)
Currently, an ongoing phase I/II dose-escalation study (NCT04506567) is evaluating fractionated 225Ac-J591 for progressive mCRPC in patients with prior treatment with 177Lu-PSMA, an ARPI, and chemotherapy (or refused/ineligible). Treatment will be given in a single fractionated cycle of 225Ac-J591 administered on days 1 and 15. The phase I component is a 3+3 dose-escalation study design with up to 18 patients, with the goal of identifying maximal tolerated dose. The phase II component will include up to 16-19 patients, with outcomes including:
- Radiographic response by PCWG3-modified RECIST 1.1 criteria and PSMA PET
- Biochemical and radiographic progression-free survival
- Circulating tumor cell counts
- Overall survival
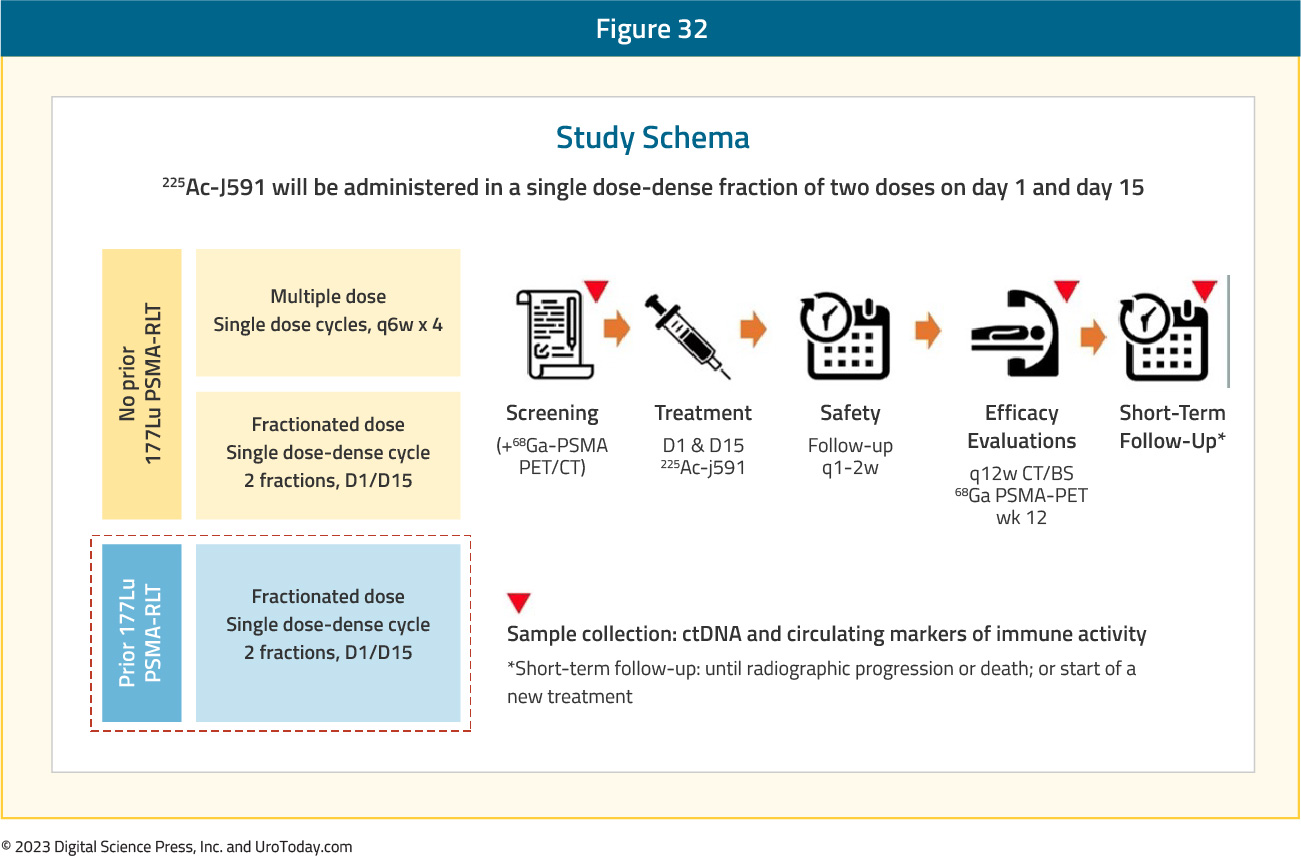
This trial is currently ongoing with enrolment having begun in August 2022.
Conclusions
We continue to witness the emergence of novel targeted therapies and drug combinations in the mCRPC disease space that aim to overcome drug resistance mechanisms and/or enhance efficacy/safety outcomes compared to currently approved agents, including ARPIs, chemotherapy, and Lu-PSMA. While not all such drugs will ultimately attain regulatory approval and become available for routine use in clinical practice, the ongoing drug development in the mCRPC space will be critical for improving survival outcomes in this high-risk population.
Related Content: New Pathways for Treating Metastatic Castration-Resistant Prostate Cancer (mCRPC)
Published November 2023
Part of an Independent Medical Education Initiative Supported by LOXO@Lilly