Thus, there remains a need for the development of novel therapeutic approaches. As summarized below, these have included immunotherapy and targeted therapies, which are discussed in this Center of Excellence mCRPC article.
IMMUNOTHERAPY
Sipuleucel-TIn 2010, Sipuleucel-T was approved in mCRPC patients in the first line setting. Sipuleucel-T is an active cellular immunotherapy, a type of therapeutic cancer vaccine, consisting of autologous peripheral-blood mononuclear cells, including antigen-presenting cells, that have been activated ex vivo with a recombinant fusion protein (PA2024). PA2024 consists of a prostate antigen, prostatic acid phosphatase, that is fused to granulocyte–macrophage colony-stimulating factor, an immune-cell activator.1
The IMPACT trial was a phase III double-blind, placebo-controlled trial that randomized 512 patients in a 2:1 fashion to either sipuleucel-T or placebo, administered intravenously every two weeks, for a total of three infusions. This trial met its primary endpoint with a 22% relative reduction in overall survival (OS) (HR 0.78, 95% CI 0.61 to 0.98). This reduction represented a 4.1-month improvement in median survival (25.8 months in the sipuleucel-T group vs. 21.7 months in the placebo group):
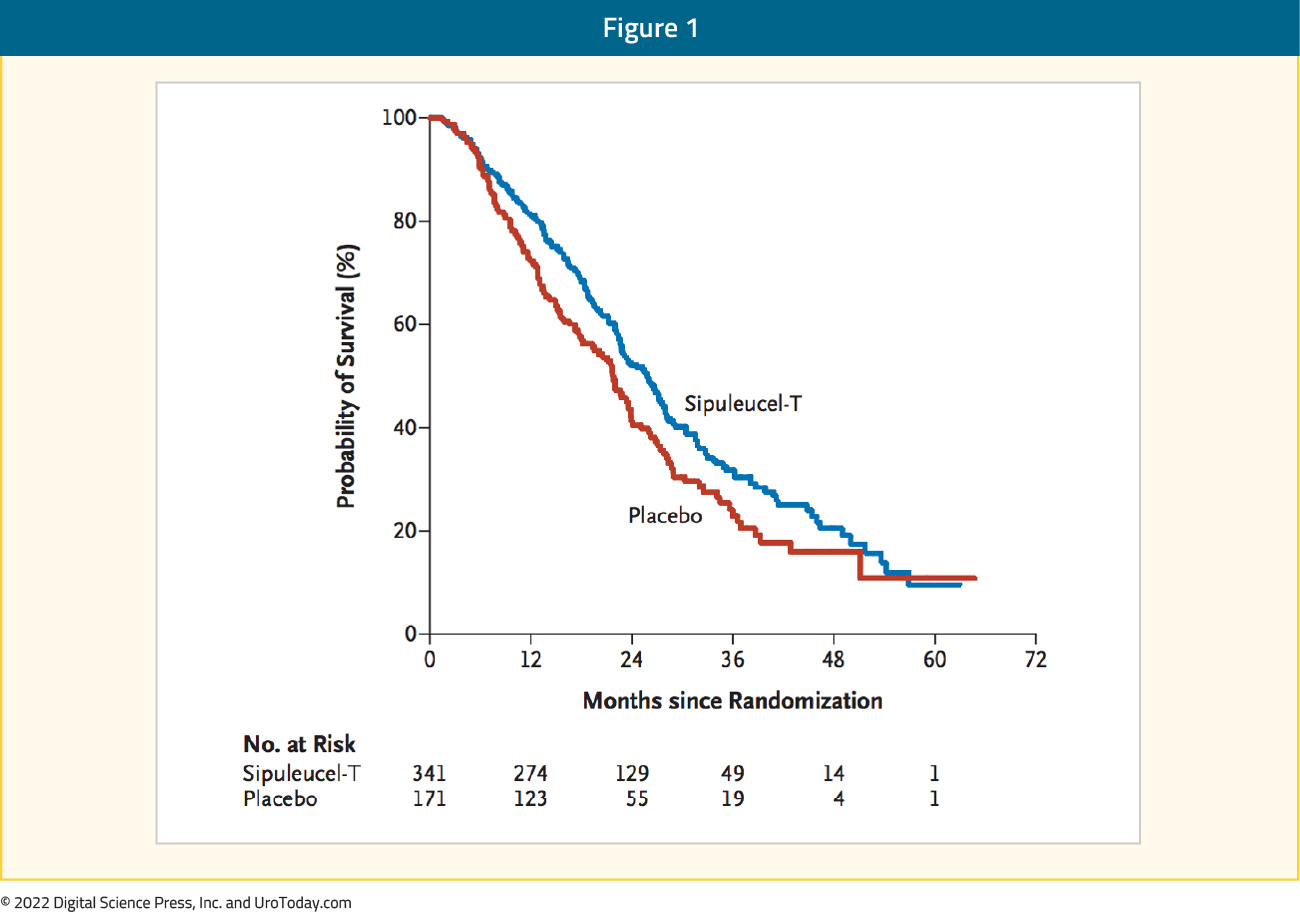
These survival benefits were not reflected in an improvement in median time to objective disease progression (3.7 versus 3.6 months) or time to clinical disease progression (HR 0.92, 95% CI 0.75 – 1.12). PSA reductions of at least 50% were seen in only 2.6% of patients in the sipuleucel-T arm (1.3% in control arm). There were no significant differences in the rates of overall or Grade 3-5 adverse events (AEs), with immune-mediated AEs such as chills and pyrexia present more commonly seen in the treatment arm.18
IMMUNE CHECKPOINT INHIBITION
Immune checkpoint inhibitors have demonstrated significant benefits in many cancer types. It is therefore not surprising that they have been tested in the advanced prostate cancer setting.Pembrolizumab
Despite promise in many tumor types, studies of immunotherapy utilizing checkpoint inhibitors have unfortunately proven generally unsuccessful in patients with advanced prostate cancer. However, in 2017, pembrolizumab was approved by the FDA for use in adult and pediatric patients with unresectable or metastatic, microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMRT) solid tumors that have progressed following prior treatment. This was the FDA’s first tissue/site-agnostic approval. This approval was based on results from a single arm trial of 149 patients with MSI-H or dMMR cancers. Of note, prostate cancer patients accounted for only two (1.3%) patients. Patients received either pembrolizumab, 200 mg every 3 weeks, or pembrolizumab, 10 mg/kg every 2 weeks. The objective response rate (ORR) in this trial was 39.6% (95% CI 31.7 to 47.9) and responses lasted six months or more for 78% of those who responded to pembrolizumab. The ORR was similar irrespective of whether patients were diagnosed with colorectal cancer (36%) or a different cancer type (46% across the 14 other cancer types).2 Notably, MSI-high status has been reported in the range of 2.2-12% of patients with advanced prostate cancer.3,4 As a result, the NCCN guidelines recommend MSI testing for men with mCRPC and consideration of pembrolizumab therapy for men with refractory MSI-high mCRPC.5
The KEYNOTE-199 trial evaluated pembrolizumab monotherapy for docetaxel-pretreated mCRPC patients. ORR was modest in this trial ranging between 3% and 5%, irrespective of PD-L1 status (cohorts 1 and 2).6 Response rates were superior in KEYNOTE-199 cohort 4, which included chemotherapy-naïve mCRPC patients who progressed on enzalutamide after initial response. These patients received pembrolizumab + enzalutamide with an ORR 12.3% (median duration of response: 8.1 months) and median rPFS of 4.2 months.
Ipilimumab + Nivolumab
Although dual immune checkpoint inhibition treatment regimens have become a standard of care treatment choice in the first line treatment of metastatic clear cell renal cell carcinoma and other tumor sites, to date such combinations have proven less effective in the mCRPC setting. The IMPACT trial, cohort A evaluated the combination of ipilimumab and nivolumab in CDK12 inactivated or mutated mCRPC cancer patients. CDK12 inactivation represents a unique subset of mCRPC with evidence of excessive tandem duplications, genomic instability, gene fusion-caused putative neoantigens, and increased tumor T cell infiltration, increasing potential susceptibility to immunotherapy. Among 28 patients, PSA ≥30% and ≥50% declines were seen in 21% and 14.2% of patients, respectively. Conversely, 21.4% experienced a rapid PSA increase by ≥ 10-fold over baseline. In terms of toxicity, grade ≥3 possible/probable/definite adverse events were noted in 25% and serious adverse events were noted in 36%.
TARGETED THERAPY
PARP Inhibitor MonotherapyWhile cytotoxic chemotherapy and androgen axis targeting agents have demonstrated benefit, emerging targeted therapeutic options have gained increasing prominence. Although underlying dominant driving mutations are not widespread in prostate cancer, there are a number of key genomic mutations that have been consistently identified in prostate cancer patients across the disease spectrum. These include gene fusion/chromosomal rearrangements (TMPRSS2-ERG), AR amplification, inactivation of tumor suppressor genes (PTEN/PI3-K/AKT/mTOR, TP53, Rb1) and oncogene activation (c-MYC, RAS-RAF).7 In the landmark analysis of 692 men with metastatic prostate cancer by Pritchard et al. published in The New England Journal of Medicine, germline DNA-repair gene mutations were present in 11.8% of metastatic prostate cancer patients, which was significantly higher than that seen in patients with localized disease (4.6%). Mutations were found in: BRCA2 (5.3%), ATM (1.6%), CHEK2 (1.9%), BRCA1 (0.9%), RAD51D (0.4%), and PALB2 (0.4%).8 Defects in DNA repair (DDR mutations) appear to be central in increasing one’s susceptibility to malignant transformation. The poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP) enzyme and BRCA 1/2 (BReast CAncer gene 1 and 2) and ATM (Ataxia-Telangiesctasia Mutated) gene products play important roles in this process.9 In patients with DDR mutations, the FDA has approved two PARP inhibitors, rucaparib and olaparib.
Rucaparib: FDA Approved in 2020 (TRITON2 trial)
The first PARP inhibitor to be approved by the FDA was rucaparib. On May 15, 2020, rucaparib was granted accelerated approval for patients with mCRPC and BRCA mutations (germline or somatic) who had progressed following treatment with androgen-axis targeted treatment and taxane-based chemotherapy, based on the phase II TRITON2 trial. TRITON2 is an international, multi-center, open-label phase II study that enrolled men with mCRPC who had disease progression following an androgen axis inhibitor, at least one taxane-based chemotherapy, and one of 13 associated homologous recombination repair gene alterations. Analysis limited to 115 patients with a BRCA alteration (median follow up 13.7 months) demonstrated an ORR of 43.5% per independent radiology review and a confirmed PSA response rate of 54.8%. Notably, ORRs were similar for patients regardless of whether a BRCA1/2 alteration was present and whether the mutation was germline or somatic. PSA response rate was higher in patients with a BRCA2 alteration.10 A separate report limited to 78 patients with non-BRCA DDR gene alterations however demonstrated limited response rates for rucaparib in this setting.11 The FDA approval of rucaparib is contingent upon ongoing assessment in confirmatory trials, namely the phase III TRITON3 trial, which is randomizing patients with mCRPC and mutations in BRCA1, BRCA2, or ATM to rucaparib or investigators choice of abiraterone, enzalutamide, or docetaxel following progression on an androgen axis inhibitor.
Olaparib: FDA Approved in 2020 (PROfound2 trial)
Shortly after the approval of rucaparib, on May 19, 2020, the U.S. Food and Drug Administration approved olaparib for the treatment of patients with germline or somatic homologous recombination repair gene-mutated mCRPC who progressed following treatment with enzalutamide or abiraterone acetate, on the basis of the phase III PROfound randomized controlled trial. To be eligible for inclusion, men must have had alterations in one of 15 pre-specified genes involved in homologous recombination repair (BRCA 1/2, ATM, BRIP1, BARD1, CDK12, CHEK 1/2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, RAD54L). The trialists then used biomarker driven stratification to derive two study cohorts: Cohort A had alterations in BRCA1, BRCA2, or ATM while Cohort B had alterations in any of the other 15 included genes. In both cohorts, patients were randomized 2:1 to olaparib versus abiraterone or enzalutamide. There was significantly improved PFS in patients with mutations of BRCA1, BRCA2, or ATM (HR 0.34, 95% CI 0.25 to 0.47):
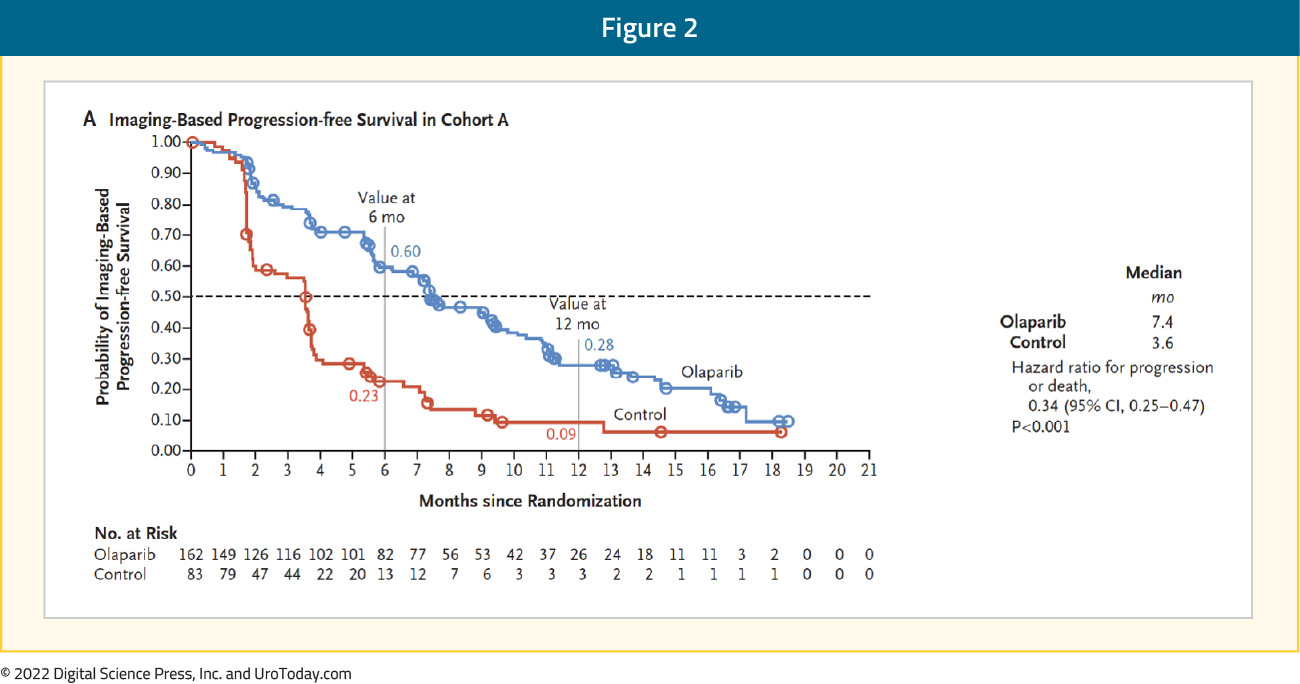
Similar, though somewhat attenuated, results were seen in the combined cohort (HR 0.49, 95% CI 0.38 to 0.63):
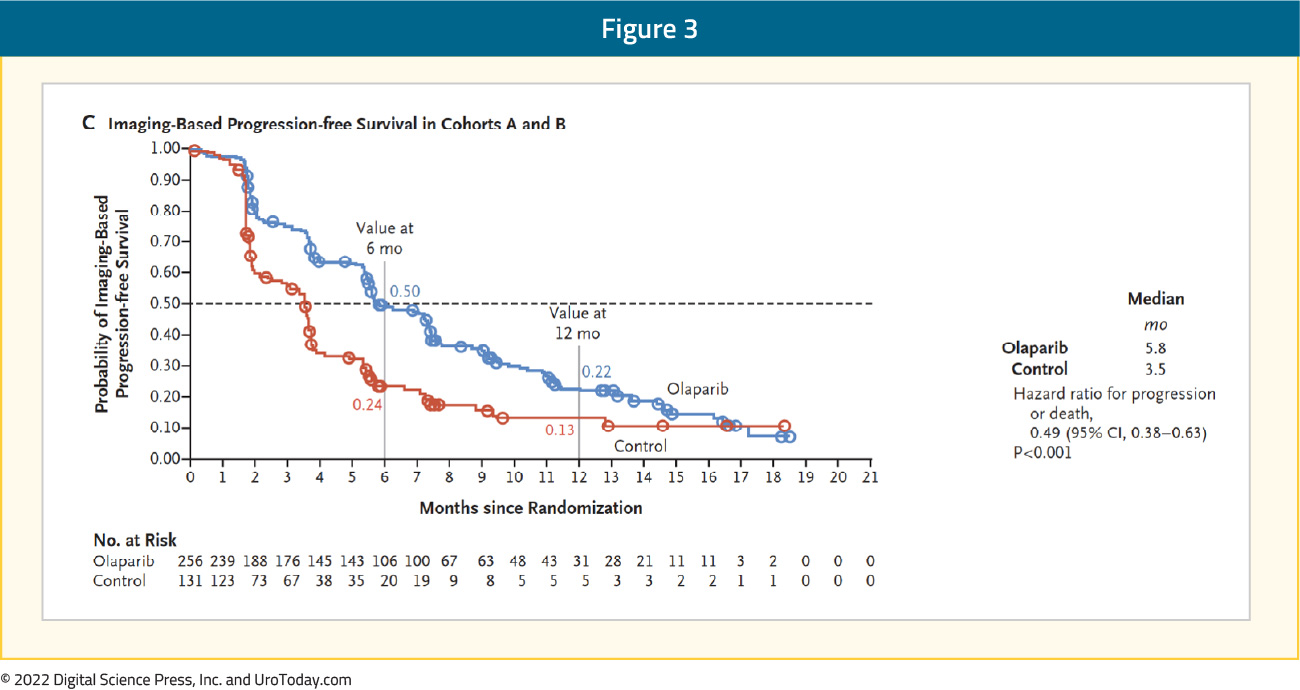
Subgroup analysis showed similar results when stratified according to prior taxane use, measurable disease at baseline, location of metastasis (bone only, visceral, or other), performance status, age at randomization, region, and baseline PSA (dichotomized around the median). In updated results, also published in 2020, treatment with olaparib demonstrated improved overall survival in Cohort A (HR 0.69, 95% CI: 0.50 to 0.97) with OS in Cohort A improving from 14.7 to 19.1 months. Notably, 84% of patients with imaging-based disease progression had crossed over from the control arm to Olaparib at time of analysis. In a cross-over adjusted sensitivity analysis, a larger effect was seen (in Cohort A: HR 0.42, 95% CI 0.19 to 0.91). In cohort B, there was no significant improvement in overall survival (17.3 and 14.0 months).12
AEs were common in both patients on olaparib (any = 95%, grade ≥ 3 = 51%) and in the control group (any = 88%, grade ≥ 3 = 38%).13 Ad hoc analysis of the PROfound trial further demonstrated that olaparib use was associated with improved median time to pain progression, opiates initiation, and improved Functional Assessment of Cancer Therapy-Prostate (FACT-P) scores.14
Talazoparib
The open-label, phase II TALAPRO-1 trial evaluated talazoparib 1 mg/day in patients with evidence of progressive mCRPC who had measurable soft-tissue disease and evidence of one of 11 DDR mutations who had progressed following taxane-based chemotherapy and abiraterone and/or enzalutamide. The primary endpoint was confirmed ORR. There were 128 patients enrolled, of whom 127 received at least one dose of talazoparib (safety population) and 104 had measurable soft-tissue disease (antitumor activity population). After a median follow-up of 16.4 months, the ORR was 29.8% (95% CI: 21.2 to 39.6%). The most common grade 3–4 treatment-emergent AEs were anemia (31%), thrombocytopenia (9%), and neutropenia (8%).15
Akt Inhibitors
Akt is a cytosolic serine/threonine kinase that has three isoforms (Akt 1, 2, and 3). It is involved in promoting growth and survival in response to extracellular stimuli and plays a critical role in regulating diverse cellular functions including cell metabolism, proliferation, apoptosis suppression, and angiogenesis. Its upregulation has been reported in a variety of human malignancies, including prostate cancer. Emerging evidence from in vitro and animal model studies have demonstrated that Akt, through the PI3K-Akt-mTOR pathway, plays a critical role in prostate cancer development and progression, with pathway deregulation associated with higher grade, advanced stage, and progression to castration-resistance. This PI3K-Akt-mTOR pathway is frequently deregulated in mCRPC and is commonly associated with tumor aggressiveness and resistance to chemotherapy or radiotherapy.16
PTEN is a protein phosphatase that has been shown to negatively regulate this pathway with PTEN loss occurring in up to 50% of mCRPC patients. Earlier efforts with drugs such as Rapamycin, Temsirolimus, and Everolimus targeting Mammalian target of rapamycin (mTOR), a downstream member of the Akt pathway, have proven unsuccessful in the treatment of prostate cancer. However, Akt isoform inhibitors, Ipatasertib and Capivasertib, have recently been evaluated in the mCRPC setting.16
Ipatasertib
Ipatasertib, an oral inhibitor of all three Akt isoforms, was evaluated in a phase II trial comparing Ipatasertib versus placebo in abiraterone-treated mCRPC patients who had previously received docetaxel. Patients with and without PTEN loss were included. The study included three arms: Ipatasertib 200 mg, Ipatasertib 400 mg, and placebo. Ipatasertib 400 mg (versus placebo) showed a trend to increased rPFS (median 8.2 vs. 6.4 months; HR 0.75, p = 0.17) and increased OS (median 18.9 months vs. 15.6 months; HR 0.72, p = 0.22). Patients with PTEN loss had a superior rPFS benefit versus those without. AEs included nausea, vomiting, diarrhea, rash, asthenia, hyperglycemia and decreased appetite. Grade ≥3 AEs were seen in 50.6%, 64.3%, and 35.4% of patients in the Ipatasertib 200 mg, Ipatasertib 400 mg, and placebo groups, respectively.17
IPATential150 is the first phase III trial to evaluate an AKT inhibitor. This trial included 1,101 previously untreated, asymptomatic, or minimally symptomatic mCRPC patients with progressive disease. Patients were randomized 1:1 to receive ipatasertib (400 mg once daily orally) plus abiraterone (1000 mg once daily orally) and prednisolone (5 mg twice a day orally) or placebo plus abiraterone and prednisolone (with the same dosing schedule). Median follow up was 19 months. In the group of patients with PTEN loss (47% of total cohort), median rPFS was superior in the ipatasertib arm (18.5 versus 16.5 months; HR 0.77, 95% CI 0.61 to 0.98):

Median PFS was non-significantly different between the two arms (19.2 months in treatment arm versus 16.6 months in the control arm; HR 0.84, 95% CI 0.71 to 0.99, p=0.043; not significant at α=0·01). Grade ≥3 AEs were seen in 70% of patients in the ipatasertib arm versus 39% in the control arm.18
Capivasertib
ProCAID is a randomized, double blind, placebo-controlled phase II study that compared capivasertib with docetaxel versus docetaxel with placebo. Capivasertib is a potent selective inhibitor of all three AKT isoforms (AKT1/2/3). Patients received up to ten 21-day cycles of docetaxel (75 mg/m2 the first day) and prednisolone (5 mg twice daily, orally, from day 1 to day 21) and were randomly assigned to receive either capivasertib 320 mg (orally, twice daily), or placebo, until the progression of the disease. The primary outcome was composite progression-free survival (cPFS) that included PSA progression event. Capivasertib addition was not associated with improved cPFS (median 7.0 vs. 6.7 months; HR 0.92, p = 0.32) irrespective of PI3K-AKT-PTEN pathway activation status. However, there was improved OS with capivasertib vs. placebo (median 31.2 vs. 20.3 months; HR = 0.54, p = 0.01).19 Updated results presented at ASCO GU 2022 demonstrated persistent OS benefits with capivasertib addition (median OS 25.3 versus 20.3 months; HR: 0.70, 95% CI: 0.47 – 1.05):
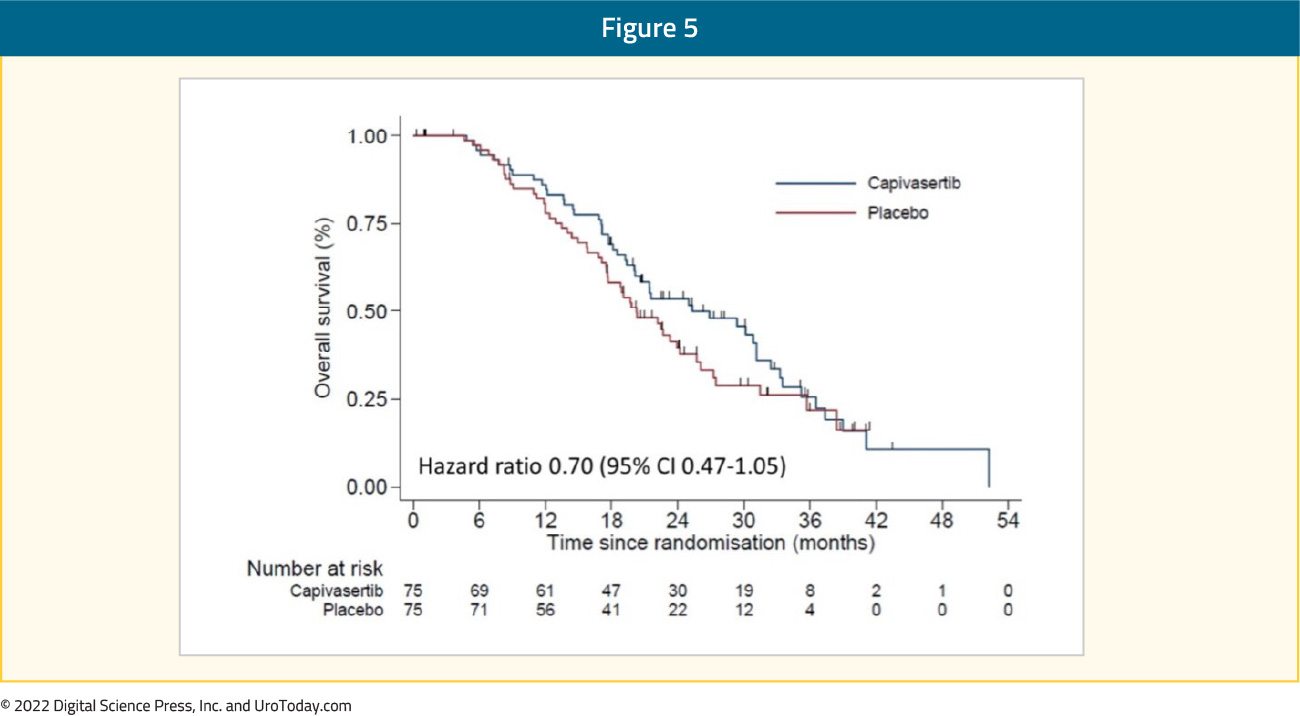
Conclusion
Immunotherapy and targeted therapies have changed the landscape of several advanced malignancies including non-small cell lung cancer, melanoma, renal cell carcinoma, and ovarian cancer, to name a few. In advanced prostate cancer, the immunotherapy efficacy has been less enthusiastic to date, however ongoing trials utilizing combination options remain promising. Targeted therapies, particularly the PARP inhibitors, have gained FDA approval and have been inserted into the mCRPC treatment armamentarium. Taken together, using immunotherapy regimens and PARP inhibitors in combination with other agents is likely the way forward and will be discussed in the Center of Excellence article discussing combination regimens.Published September 2022
Written by:
- Rashid Sayyid, MD MSc, University of Toronto, Toronto, ON
- Zachary Klaassen, MD MSc, Medical College of Georgia, Augusta, Georgia, USA