Introduction: Despite the approval of numerous agents in this setting, patients with metastatic castrate-resistant prostate cancer (mCRPC) have a poor prognosis, with an estimated median overall survival (OS) of approximately three years with currently approved first-line agents.1-3
In the “real world”, survival outcomes are even worse, with an estimated median OS of less than two years, likely due to patients in the “real world” often never receiving second- and third-line treatment regimens.4 As such, it is clear that new agents and/or combinations in this setting are sorely needed.
Poly (adenosine diphosphate-ribose) polymerase inhibitors (PARPi) prevent the repair of DNA single-stranded breaks and promote their conversion to double-stranded breaks. These agents have demonstrated OS benefits in the second-line line setting for HRR-mutated (HRRm) mCRPC patients, who had progressed on prior androgen receptor signaling inhibitors (ARSI).5-7 Preclinical models have suggested synergistic mechanisms of action for PARPi and ARSIs. PARPi upregulates androgen receptor signaling, enhancing ARSI activity. Conversely, ARSIs inhibit the transcription of some HRR genes, inducing an HRR deficiency-like state, thus potentiating PARPi activity.8-10 These mechanisms of action suggest that the PARPi/ARSI combination may have clinical efficacy, irrespective of HRR mutational status. This was further supported by the results of a phase II trial of combination olaparib + abiraterone acetate that demonstrated improved radiographic progression-free survival (rPFS) in docetaxel pre-treated, HRR biomarker unselected mCRPC patients.11
Over the past year, three randomized clinical trials evaluating the combination of a PARPi + an ARSI in the first line mCRPC treatment setting have been presented/published. In this Center of Excellence article, we will summarize and discuss the latest evidence from PROpel, MAGNITUDE, and TALAPRO-2.
PROpel
Originally presented at ASCO GU 2022 and since published in NEJM Evidence, PROpel is a global, randomized, double-blind phase 3 trial of abiraterone and olaparib versus abiraterone and placebo in patients with mCRPC treated in the first-line setting. Patients in PROpel were enrolled irrespective of HRRm status, ascertained via circulating tumor DNA (ctDNA) or tissue testing. Patients were randomized (1:1) to receive abiraterone (1000 mg once daily) plus prednisone/prednisolone with either full dose olaparib (300 mg twice daily) or placebo. Randomization was stratified by site of distant metastases and by prior history of taxane use in the metastatic hormone-sensitive state. Prior abiraterone use was not permitted, with other ARSIs permitted if discontinued ≥12 months prior to study enrollment. The primary end point was imaging-based progression-free survival (ibPFS) by investigator assessment. Overall survival was among the secondary end points. The trial design for PROpel is as follows:

PROpel randomized 399 and 397 patients to the PARPi + ARSI and placebo + ARSI arms, respectively. Median age was 70 years, with a median PSA of 17 – 18 ng/ml, and approximately 25% had received prior docetaxel. The most frequent site of distant metastases was the bone (85- 88%). HRRm status was positive in 28% and 29% of patients in the intervention and control arms, respectively, with HRRm status unknown in 2.3%. BRCA1/2 mutations were observed in 11.8% and 9.6% of patients, respectively.
At a median follow-up of 19.4 months, the combination of olaparib + abiraterone significantly improved median investigator-assessed rPFS from 16.6 to 24.8 months (HR: 0.66, 95% CI: 0.54 – 0.81, p<0.001). Consistent results were observed when rPFS was assessed via blinded-independent central review (BICR):
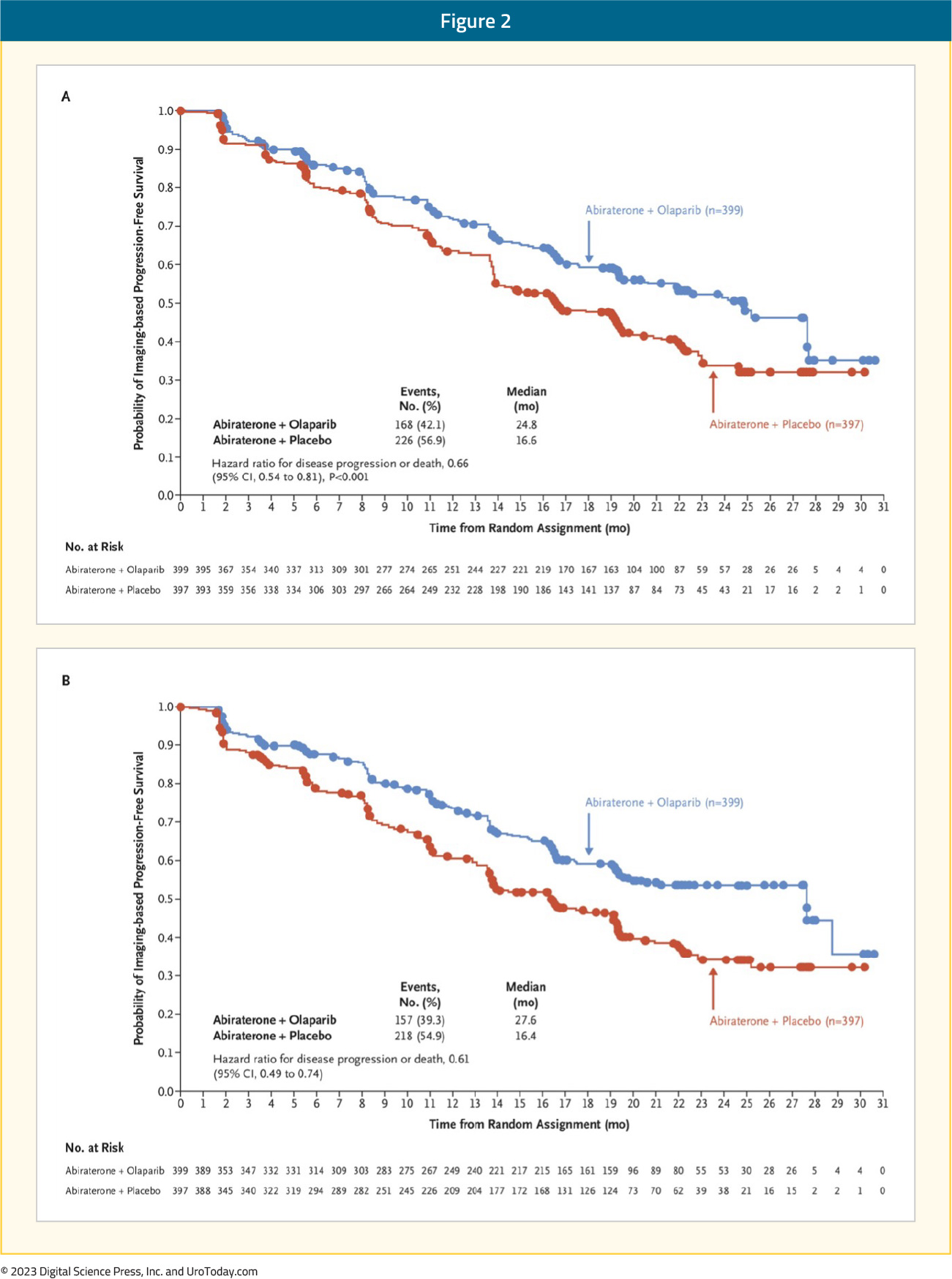
rPFS benefits were observed irrespective of HRRm status, although, as expected, the magnitude of effect was higher in the HRRm (HR: 0.50, 95% CI: 0.34 – 0.73) versus non-HRRm patients (HR: 0.76, 95% CI: 0.60 – 0.97). Additionally, on subgroup analysis, rPFS benefits were observed irrespective of prior docetaxel exposure and metastatic site. At this first data cut-off, OS data maturity was only 28.6%. There was a non-significant 14% improvement in OS mortality rate in the combination arm (HR: 0.86, 95 CI: 0.61 – 0.90):

Other secondary endpoints, including time to first subsequent therapy and time to second progression also favored the addition of olaparib to abiraterone.
The percentage of patients with any treatment-emergent adverse event (TEAE) or death was similar across both arms (97% and 95%). A higher proportion of patients receiving abiraterone + olaparib (47%) had grade ≥3 AEs, compared to those receiving abiraterone plus placebo (38%). The most common AE was anemia, occurring in 46% of patients receiving olaparib + abiraterone, compared to 16% of patients receiving placebo + abiraterone, with grade ≥3 anemia occurring in 15% and 3% of patients, respectively:

Quality of life, assessed via the Functional Assessment of Cancer Therapy – Prostate questionnaire, was similar in both arms.12
The pre-planned final OS analysis was presented at ASCO GU 2023, with a consistent trend towards an OS benefit with combination olaparib + abiraterone versus placebo + abiraterone in the overall intention-to-treat (ITT) population (data maturity: 47.9%; HR 0.81, 95% CI: 0.67 – 1.00, p=0.054), with median OS of 42.1 and 34.7 months, respectively:
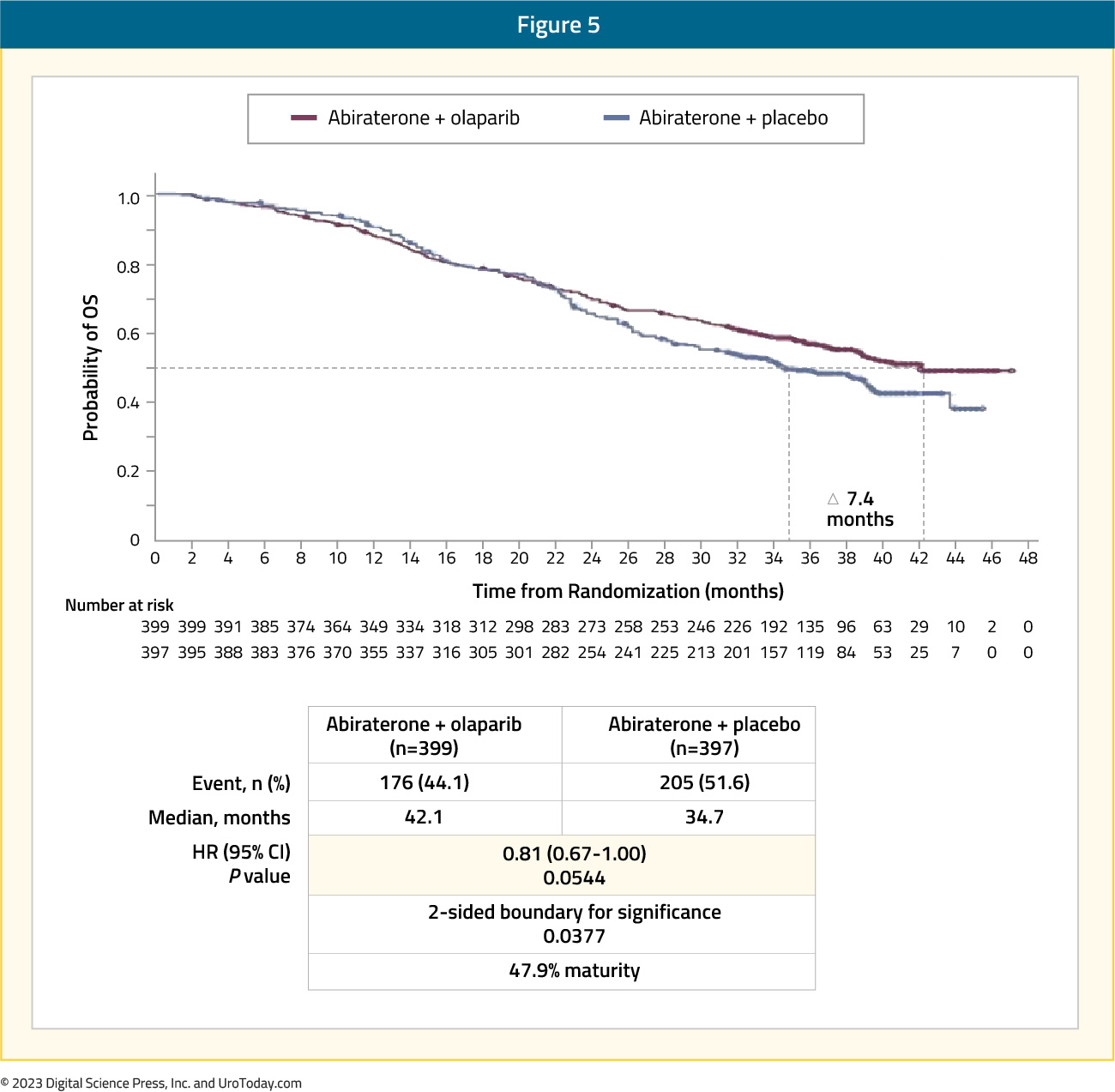
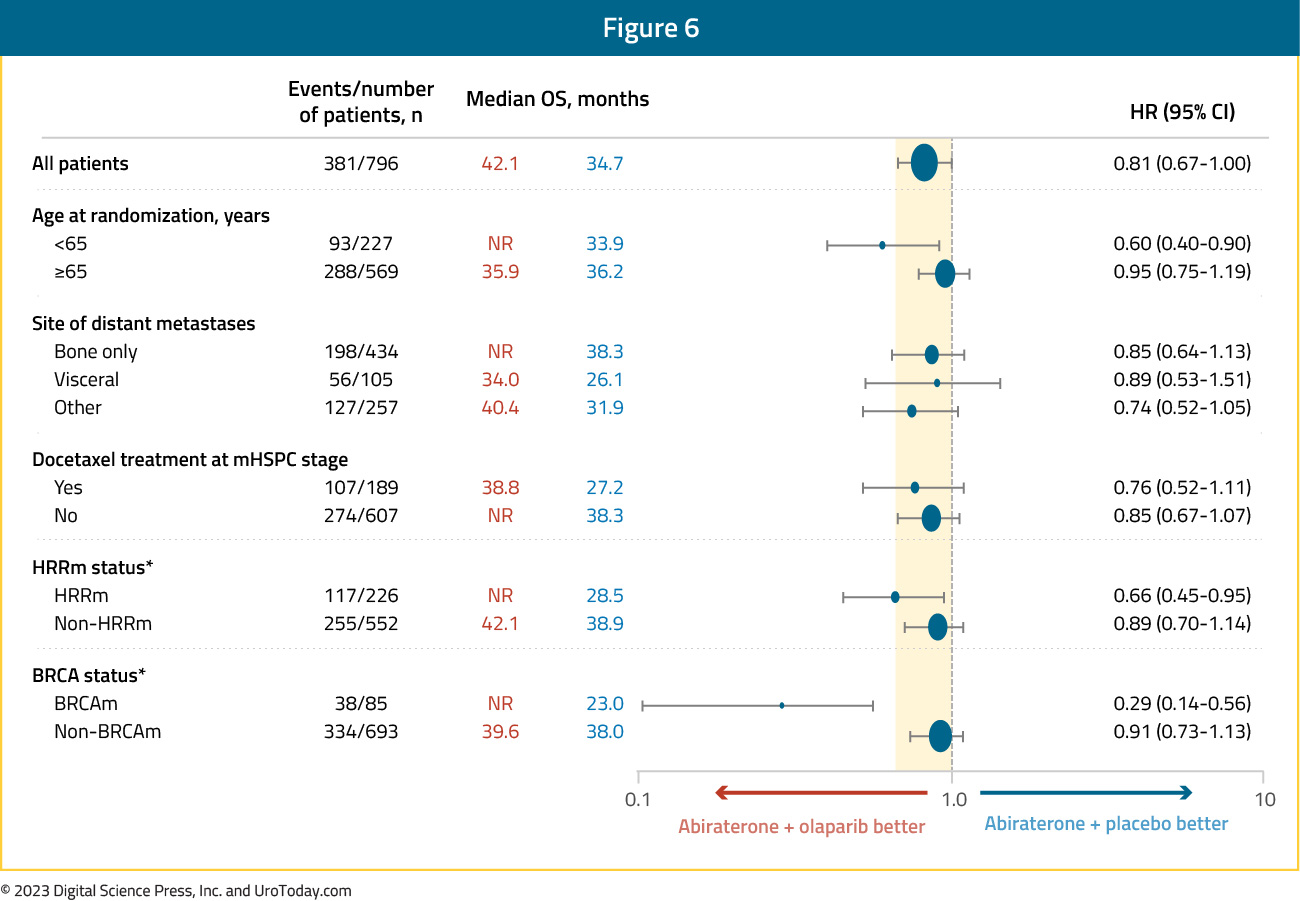
Subgroup analysis demonstrated non-significant OS benefits irrespective of HRRm status, with a larger magnitude of effect observed in the HRRm subgroup (HR: 0.66 versus 0.89 for non-HRRm). Although the number of BRCA-mutated patients in this cohort was limited (n=85), there appears to be a clinically meaningful OS benefit in this subgroup (HR: 0.29, 95% CI: 0.14 – 0.56):
No new safety signals were observed with this updated report.
At the Oncologic Drugs Advisory Committee (ODAC) meeting on April 28, 2023, the FDA made several detrimental remarks regarding the PROpel data:
- The “FDA is concerned that efficacy and safety have not been demonstrated outside of the small population of patients with tumor BRCA mutations, and that the addition of olaparib to abiraterone may cause harm in patients who are definitively negative for tumor BRCA mutations”
- “For patients with non-BRCAm, in whom olaparib is unlikely to be effective, the addition of olaparib may represent prolonged exposure to a toxic placebo as the efficacy of abiraterone may effectively mask an ineffective add-on therapy”
- “Given the FDA’s concern for harm from adding olaparib to abiraterone in patients without tumor BRCAm, the FDA asks the ODAC to consider whether the indication for olaparib in combination with abiraterone in mCRPC should be restricted to patients whose tumors have a BRCA mutation”
Of note, this combination is approved in the European Union.
MAGNITUDE
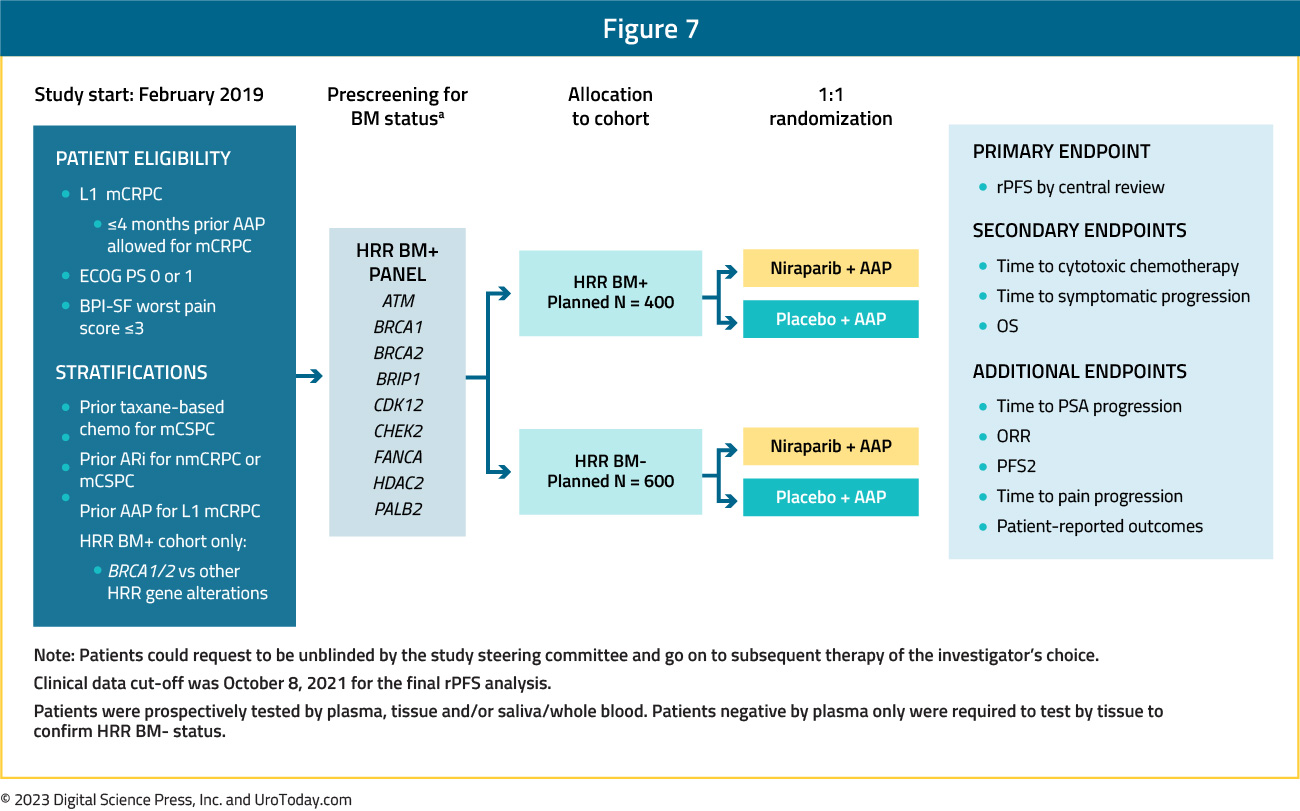
Presented concurrently with PROpel at GU ASCO 2022 and since published in Journal of Clinical Oncology,14 MAGNITUDE is a phase III, randomized, double-blind, placebo-controlled, multicenter trial that evaluated the combination of niraparib plus abiraterone acetate + prednisone in mCRPC patients receiving first-line treatment. In contrast to PROpel and TALAPRO-2, this trial included biomarker pre-screened cohorts, meaning that all patients had HRR mutational status determined prior to study enrollment. HRR mutational status was determined using tissue and/or blood samples. Patients with a gene alteration (ATM, BRCA1/2, BRIP1, CDK12, CHEK2, FANCA, HDAC2, or PALB2) detected by ≥1 assay were assigned to the HRR+ cohort, whereas those with both assays negative were included in the HRR- cohort. Patients in the HRR+ and HRR- cohorts underwent 1:1 randomization to to receive either niraparib 200 mg once daily (usual dose: 400 mg) and abiraterone acetate 1,000 mg once daily plus prednisone 5 mg twice daily or placebo + abiraterone + prednisone. Given the lower likelihood of clinical benefit with combination treatment in the HRR- cohort, a futility analysis was preplanned when approximately 200 patients had been enrolled and approximately 125 composite end point events (the first of either PSA progression, radiographic progression, or death) had occurred. For the HRR+ cohort, primary and secondary endpoints were tested using a pre-specified strategy. The primary rPFS endpoint was powered for and tested first in the BRCA1/2 subgroup and if statistical significance was reached, the remainder of the HRR+ patients would be analyzed. Approximately 50% of HRR+ patients were specified to be BRCA1/2 positive. The trial design for MAGNITUDE is as follows:
Results of the futility analysis in the HRR- cohort (n=233) demonstrated no benefit for combination niraparib + abiraterone versus placebo + abiraterone. The hazard ratio for the composite endpoint was 1.09, with futility defined as a hazard ratio greater than 1.0. As anticipated, additional grade ≥3 AEs occurred with addition of niraparib. Given the increased toxicity with no observed oncologic efficacy, the independent data monitoring committee recommended suspending enrollment in the HRR- cohort.
The HRR+ cohort included 423 patients, with 212 and 211 randomized to combination niraparib + abiraterone and placebo + abiraterone, respectively. In this cohort, 3% of patients had prior exposure to an ARSI and 23% had received <4 months of prior abiraterone for mCRPC. Median age was 69 years in both arms, and median PSA was higher in the experimental arm (21.4 versus 17.4 ng/ml). Median follow-up in the HRR+ cohort was 18.6 months. In the BRCA1/2 subgroup, median rPFS, assessed via BICR, was significantly prolonged with combination niraparib + abiraterone (16.6 versus 10.9 months; HR: 0.53, 95% CI: 0.36 – 0.79, p=0.001). A similar rPFS benefit with combination niraparib + abiraterone was next observed in the HRR+ cohort (16.5 versus 13.7 months; HR: 0.73, 95% CI: 0.56 – 0.96, p=0.022):
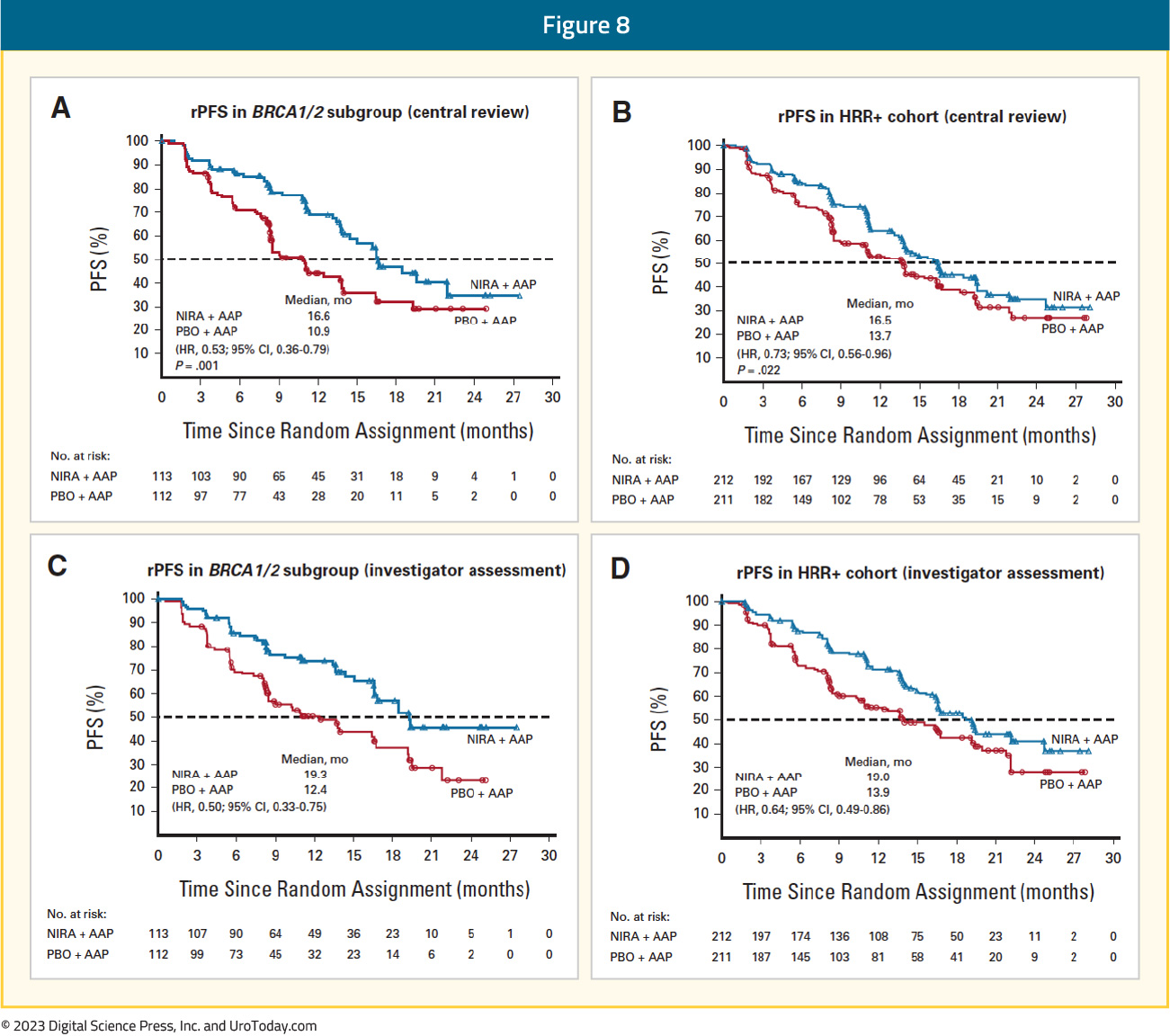
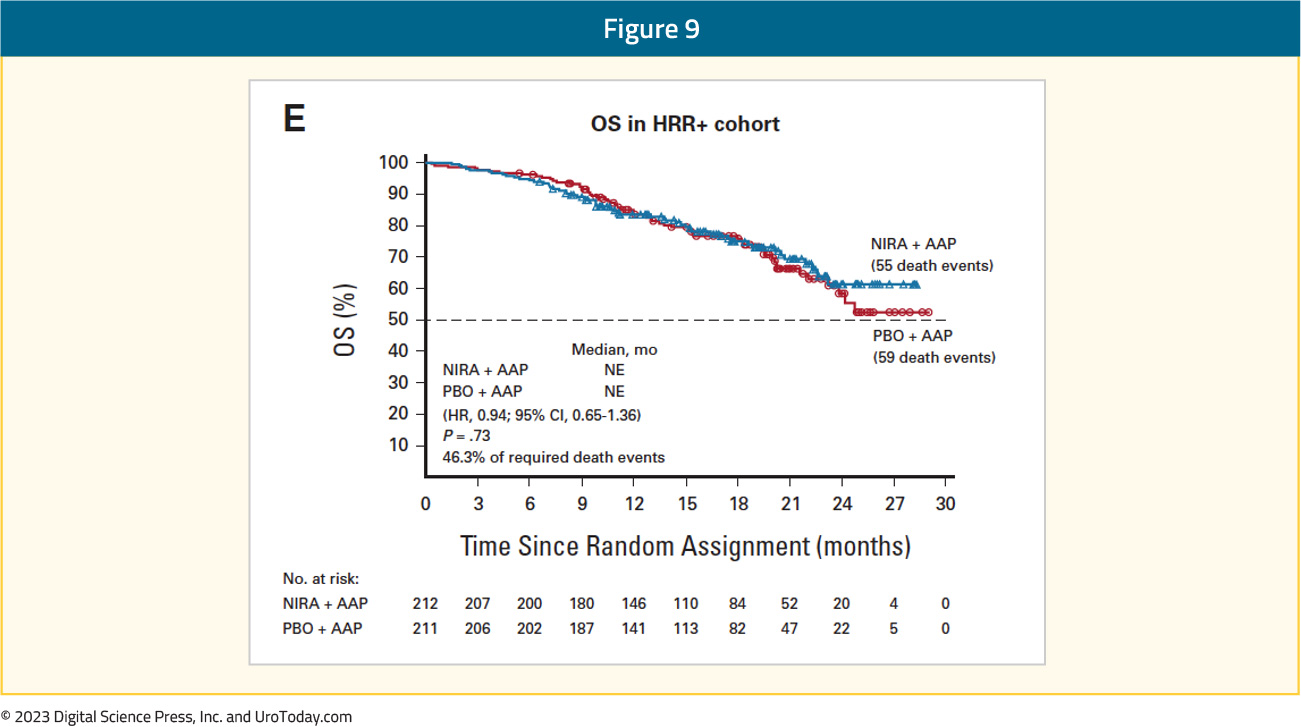
At the current time, OS data remain immature, with only 46.3% of the required events for final analysis having occurred. Crude, unadjusted analysis demonstrated an OS benefit of only 6% in the HRR+ cohort (HR: 0.94, 95% CI: 0.65 – 1.36). However, pre-specified, multivariate analysis OS, adjusting for baseline variables, favored the niraparib + abiraterone arm (HR: 0.77, 95% CI: 0.53 – 1.12). A similar pattern was observed for the BRCA1/2 mutated patients, with an HR of 0.88 (95% CI: 0. 58 – 1.34) when the overall stratified HR definition was used, compared to an HR of 0.54 (95% CI: 0.33 – 0.88) when the inverse probability of censoring weighting (IPCW) definition was employed:
In the HRR+ cohort, a similar incidence of any-grade TEAEs were observed in both arms (99.1% versus 94.3%); however, grade ≥3 AEs occurred more frequently in the niraparib/abiraterone group (67% versus 46%). Grade ≥3 anemia occurred in 30% and 8% of patients in the niraparib + abiraterone and placebo + abiraterone groups, respectively.
TALAPRO-2
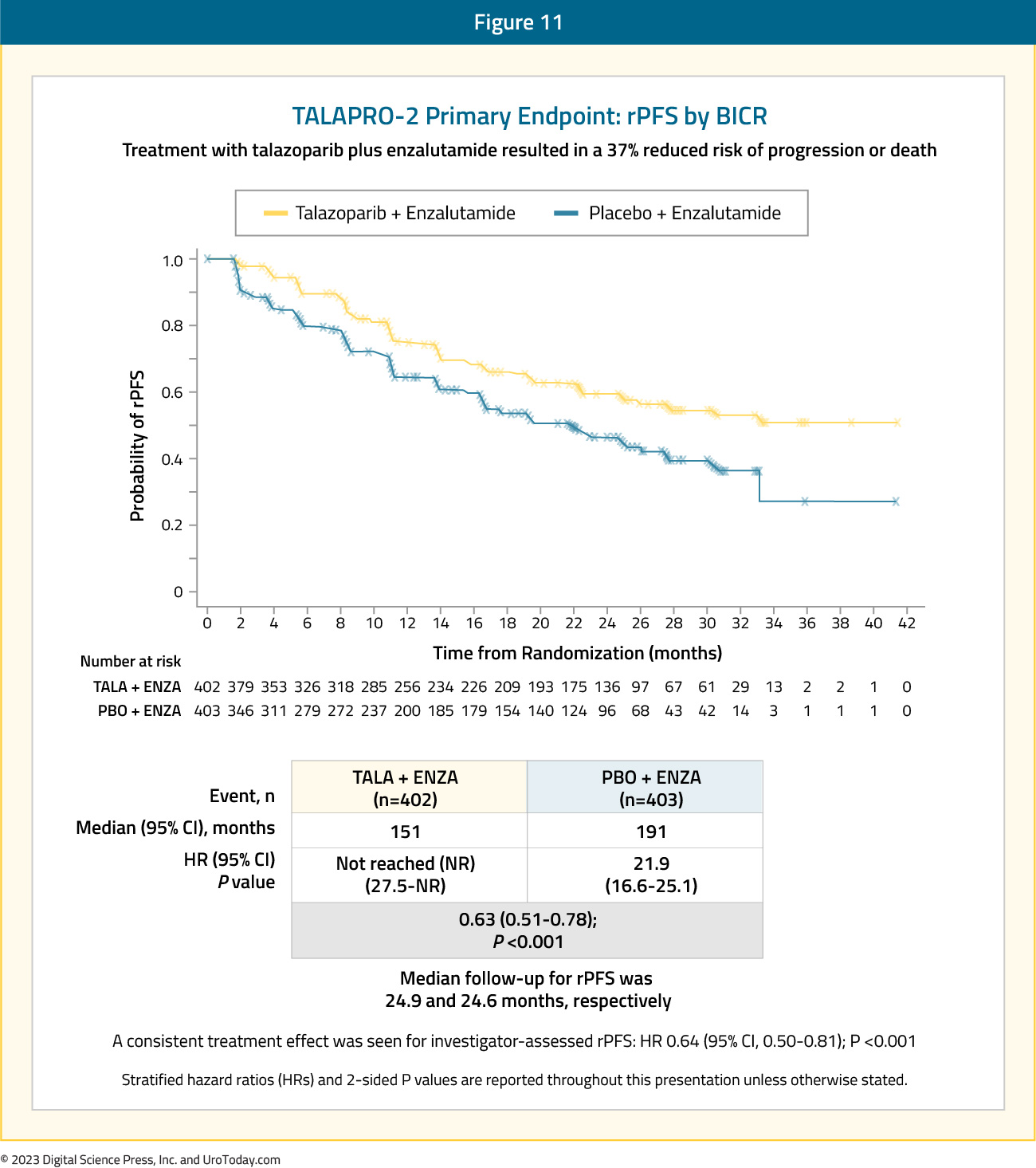
Presented at ASCO GU 2023, TALAPRO-2 is a phase III randomized, double-blind, placebo-controlled trial that evaluated the combination of talazoparib and enzalutamide in the first-line treatment setting for mCRPC patients. Patients were randomized 1:1 to talazoparib 0.5 mg once daily (reduced dose from standard of 1.0 mg) plus enzalutamide 160 mg once daily versus placebo + enzalutamide. Prior use of docetaxel and abiraterone in the mHSPC, but not in the mCRPC setting, was permitted. No prior use of an androgen receptor inhibitor was permitted. Similar to PROpel, this was a biomarker unselected cohort of ‘all comers’, although randomization was stratified by HRR gene alteration status (deficient versus non-deficient or unknown). The primary endpoint was rPFS, assessed via BICR, and OS was a key secondary endpoint. The trial design for TALAPRO-2 is as follows: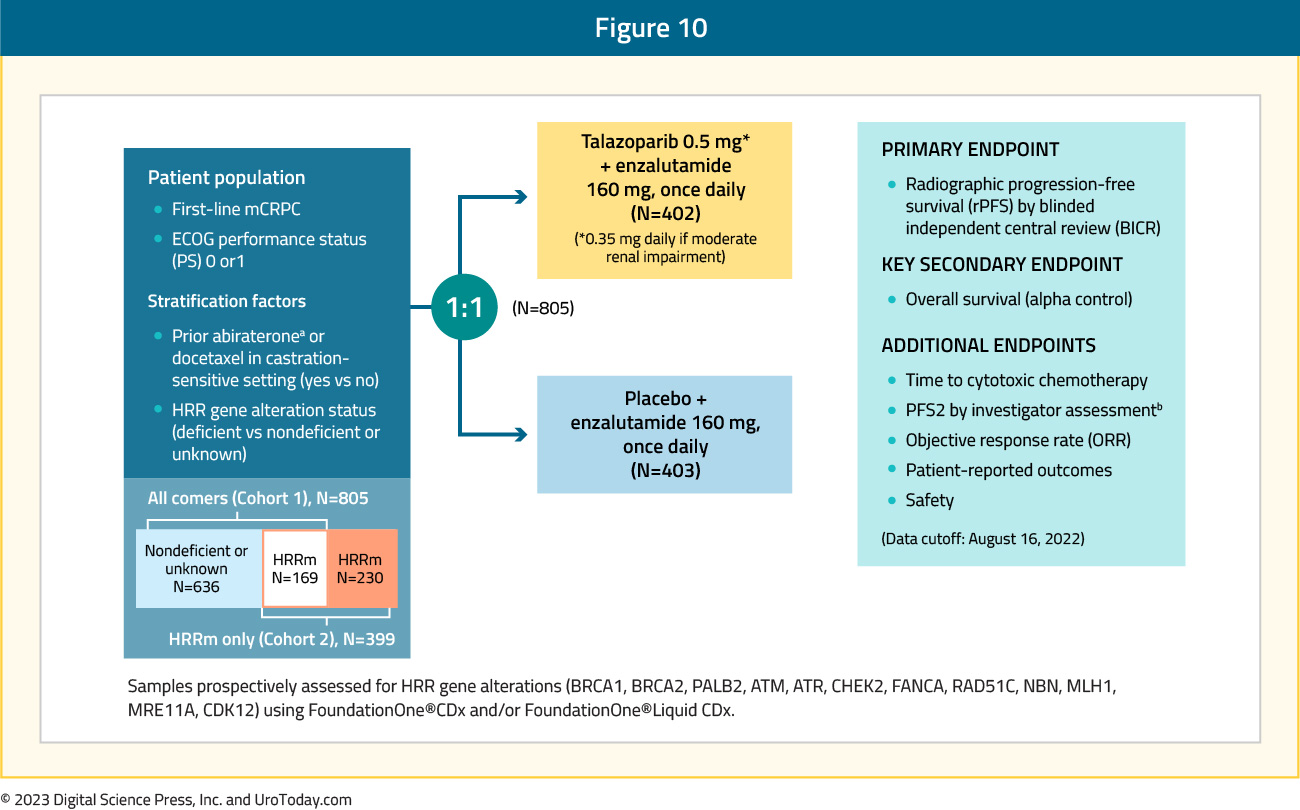
The median age was 71 years, with median serum PSA slightly higher in the intervention arm (18.2 versus 16.2 ng/ml), and 21% of patients in each arm having HRR deficient status. The combination of talazoparib + enzalutamide was associated with a 37% decrease in the rPFS hazard, compared to placebo + enzalutamide (median: not reached versus 22 months; HR: 0.63, 95% CI: 0.51 – 0.78, p<0.001):
Similar to PROpel, rPFS benefits were observed in both the HRR mutated and non-mutated subgroups. As expected, the magnitude of effect was larger in the HRR mutated (median rPFS: 28 versus 16 months; HR: 0.46, 95% CI: 0.30 – 0.70, p<0.001) versus non-mutated subgroup (HR: 0.66, 95% CI: 0.49 – 0.91, p=0.009). Similar to other trials, OS data remains immature, with a trend towards an OS improvement in the overall cohort (HR: 0.89, 95% CI: 0.69 – 1.14, p=0.35). Grade ≥3 AEs occurred more commonly in in the intervention arm (75% versus 45%), with grade ≥3 anemia observed in 46.5% and 4.2% of patients, respectively. The onset of grade 3-4 anemia occurred at a median time of 3.3 months. The most common TEAEs resulting in talazoparib dose reductions were anemia (43.2%), neutropenia (15.1%), and thrombocytopenia (5.5%).15
Discussion
Although all three trials evaluated the combination of a PARPi and an ARSI in the first line mCRPC setting, there are important differences in the study designs that may explain the observed differences. While olaparib (PROpel) inhibits PARP 1, 2, and 3, both niraparib (MAGNITUDE) and talazoparib (TALAPRO-2) inhibit PARP 1 and 2 only. Furthermore, full dose olaparib was used in PROpel, compared to reduced doses of niraparib (200 mg instead of 400 mg) and talazoparib (0.5 mg instead of usual 1.0 mg) in MAGNITUDE and TALAPRO-2, due to drug-limiting toxicities. ARSI choice also varied, with an androgen synthesis inhibitor (abiraterone) used in PROpel and MAGNITUDE versus an androgen receptor inhibitor (enzalutamide) in TALAPRO-2. With regards to study populations, PROpel and TALAPRO-2 included biomarker unselected mCRPC patients, whereas MAGNITUDE pre-screened eligible subjects for biomarker mutational status, with subsequent allocation to either a biomarker positive or negative cohort. While prior docetaxel exposure during the castrate-sensitive setting was permitted in all three trials (not within six months of enrolment in TALAPRO-2), there were significant between study differences in prior ARSI use eligibility criteria. Prior abiraterone use was not allowed in PROpel, whereas permitted in MAGNITUDE if given for ≤4 months in the first line mCRPC setting and in TALAPRO-2 if given in the castrate-sensitive state. Prior androgen receptor inhibitor use was not permitted in TALAPRO-2 but permitted in PROpel if stopped ≥12 months prior to enrollment and in MAGNITUDE, if used in the castrate-sensitive or non-metastatic castrate-resistant settings.
A meta-analysis of all three trials demonstrated that the first-line combination of a PARPi + ARSI is associated with a 35% improvement in rPFS in “all comer” mCRPC patients treated in the first-line setting (HR: 0.65, 95% CI: 0.56 – 0.76, p<0.001). This pooled analysis demonstrated significant rPFS benefits in the non-HRRm cohort (HR: 0.74, p=0.003), although, as expected, the magnitude of effect was larger in HRRm patients (HR: 0.55, p<0.001). In the overall cohort, pooled OS analysis from PROpel and TALAPRO-2 (not available from MAGNITUDE) demonstrated a 16% OS improvement in the overall cohort (HR: 0.84, 95% CI: 0.72-0.98, p=0.02). OS in the HRRm (HR: 0.79, 95% CI: 0.56-1.12) and the BRCA1/2-mutated cohorts (HR:0.53, 95% CI:0.18-1.56) were non-significantly improved, albeit with a higher effect magnitude compared to the overall cohort. With regards to TEAEs, there was a 45% increase in the relative risk of grade ≥3 events (RR: 1.45, 95% CI: 1.25-1.68, p<0.001). The relative risks of any-grade and grade ≥3 treatment-emergent anemia were increased by approximately three- and six-fold, respectively.
Conclusion
The combination of a PARPi plus an ARSI in the first-line treatment setting for mCRPC patients is associated with consistent rPFS benefits in HRRm patients (particularly those with BRCA mutations), with a less clinically significant benefit observed in non-HRRm patients in two of three published studies to date. OS data remain immature and grade ≥3 AEs, particularly anemia, remain a concern. Future follow-up of these studies will help determine the ideal patient cohorts most likely to benefit from this combination approach.Written by:
- Rashid K. Sayyid, MD, MSc, University of Toronto, Toronto, ON
- Zachary Klaassen, MD, MSc, Medical College of Georgia, Augusta, Georgia, USA