Kidney cancer is a broad, encompassing term that borders on colloquial. While most physicians are referring to renal cell carcinoma when they say “kidney cancer”, a number of other benign and malignant lesions may similarly manifest as a renal mass. Considering only the malignant causes, kidney cancers may include renal cell carcinoma, urothelium-based cancers (including urothelial carcinoma, squamous cell carcinoma, and adenocarcinoma), sarcomas, Wilms tumor, primitive neuroectodermal tumors, carcinoid tumors, hematologic cancers (including lymphoma and leukemia), and secondary cancers (i.e. metastases from other solid organ cancers).
Epidemiology
In the United States, cancers of the kidney and renal pelvis comprise the 6th most common newly diagnosed tumors in men and 10th most common in women.1 In 2018, an estimated 65,340 people will be newly diagnosed with cancers of the kidney and renal pelvis in the United States. In men, this comprises 42,680 estimated new cases in 2018 representing 5% of all newly diagnosed cancers. In women, 22,660 new cases are anticipated in 2018 representing 3% of all newly diagnosed cancers. Additionally, 14,970 people are expected to die of kidney and renal pelvis cancers in 2018 in the United States, with this being the 10th most common cause of oncologic death among men.
In Europe, results are similar. In 2018, the incidence of kidney cancer is estimated at 136,500 new cases representing 3.5% of all new cancer diagnoses.2 This corresponds to an estimated age standardized rate (ASR) of 13.3 cases per 100,000 population. As in the United States, the incidence of kidney and renal pelvis cancers is higher among men (incidence 84,9000, 4.1% of all cancers, ASR 18.6 per 100,000) than women (incidence 51,600, 2.8% of all cancers, ASR 9.0 per 100,000). Correspondingly, 54,700 people were estimated to die of kidney and renal pelvis cancers in Europe in 2018, accounting for 2.8% of all oncologic deaths. The age standardized mortality rate was 4.7 deaths per 100,000 population. Again, death from kidney and renal pelvis cancer was more common among men (mortality 35,100, 3.3% of oncologic deaths, ASR 7.1 per 100,000) than among women (mortality 19,600, 2.3% of oncologic deaths, ASR 2.7 per 100,000). Interestingly, within Europe, there is considerable variation in the incidence and mortality of kidney and renal pelvis cancer between countries.2
While the aforementioned data have already demonstrated that gender is strongly associated with the risk of both diagnosis of and death from kidney and renal pelvis cancers, age also importantly moderates this risk. Among patients in the United States, the probability of developing kidney and renal pelvis cancer rises nearly ten fold from age <50 to age >70 years.1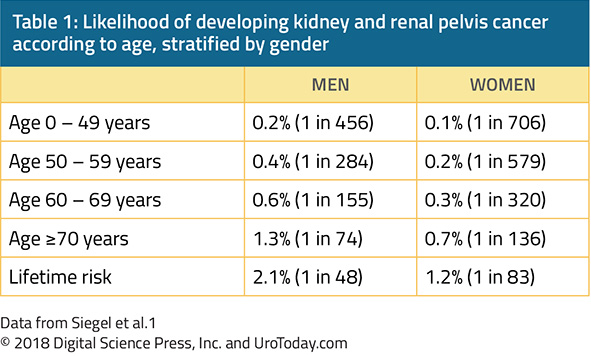
Thus, kidney cancer is predominantly a disease of older adults, with the typical presentation being between 50 and 70 years of age. However, over time, rates of diagnosis of kidney cancer have increased fastest among patients aged less than 40 years old.3
In the United States, kidney cancers are more common among African Americans, American Indians, and Alaska Native populations while rates are lower among Asian Americans.4 Worldwide, the highest rates are found in European nations while low rates are seen in African and Asian countries.4
The vast majority of patients have localized disease at the time of presentation. According to Siegel et al., 65% of all patients diagnosed with kidney and renal pelvis tumors between 2007 and 2013 had localized disease at the time of presentation while 16% had regional spread and 16% had evidence of distant, metastatic disease.1 This is in large part due to incidental diagnosis due to the increased use of ultrasonography and computed tomography in patients presenting with abdominal distress. In fact, 13 to 27% of abdominal imaging studies demonstrate incidental renal lesions unrelated to the reason for the study5 and approximately 80% of these masses are malignant.6 Dr. Welch and colleagues demonstrated elegantly that the use of computed tomography is strongly related to the likelihood of undergoing nephrectomy, likely due to detection of renal masses. Thus, with the increasing utilization of abdominal imaging, the incidence of kidney cancer has increased by approximately 3 to 4% per year since the 1970s.
Renal Cell Carcinoma
Renal cell carcinoma (RCC) is the most common kidney cancer. A number of histological subtypes have been recognized including conventional clear cell RCC (ccRCC), papillary RCC, chromophobe RCC, collecting duct carcinoma, renal medullary carcinoma, unclassified RCC, RCC associated with Xp11.2 translocations/TFE3 gene fusions, post-neuroblastoma RCC, and mucinous tubular and spindle cell carcinoma. Conventional ccRCC comprises approximately 70-80% of all RCCs while papillary RCC comprises 10-15%, chromophobe 3-5%, collecting duct carcinoma <1%, unclassified RCC 1-3%, and the remainder are very uncommon.
Histologically, most of these tumors are believed to arise from the cells of the proximal convoluted tubule given their ultrastructural similarities. Renal medullary carcinoma and collecting duct carcinoma, relatively uncommon and aggressive subtypes of RCC, are believed to arise more distally in the nephron.
Familial RCC Syndromes
While the vast majority of newly diagnosed RCCs are sporadic, hereditary RCCs account for approximately 4% of all RCCs. Due in large part to the work of Dr. Linehan and others, the understanding of the underlying molecular genetics of RCC have progressed significantly since the early 1990s. These insights have led to a better understanding of both familial and sporadic RCCs.
Von Hippel-Lindau disease is the most common cause of hereditary RCC. Due to defects in the VHL tumor suppressor gene (located at 3p25-26), this syndrome is characterized by multiple, bilateral clear cell RCCs, retinal angiomas, central nervous system hemangioblastomas, pheochromocytomas, renal and pancreatic cysts, inner ear tumors, and cystadenomas of the epididymis. RCC develops in approximately 50% of individuals with VHL disease. These tumors are characterized by an early age at the time of diagnosis, bilaterality, and multifocality. Due in large part to improved management of the CNS disorders in VHL disease, RCC is the most common cause of death in patients with VHL.
Hereditary papillary RCC (HPRCC) is, as one would expect from the name, associated with multiple, bilateral papillary RCCs. Due to an underlying constitutive activation of the c-Met proto-oncogene (located at 7q31), these tumors also present at a relatively early age. However, overall, these tumors appear in general to be less aggressive than corresponding sporadic malignancies.
In contrast, tumors arising in hereditary/familial leiomyomatosis and RCC (HLRCC), due to a defect in the fumarate hydratase (1q42-43) tumor suppressor gene, are typically unilateral, solitary, and aggressive. Histologically, these are typically type 2 papillary RCC, which has a more aggressive phenotype, or collecting duct carcinomas. Extra-renal manifestations include leiomyomas of the skin and uterus and uterine leiomyosarcomas which contribute to the name of this sydrome.
Birt-Hogg-Dube, due to defect in the tumor suppressor folliculin (17p11), is associated with multiple chromophobe RCCs, hybrid oncocytic tumors (with characteristics of both chromophobe RCC and oncocytoma), oncocytoma. Less commonly, patients with Birt-Hogg-Dube may develop clear cell RCC or papillary RCC. Non-renal manifestations include facial fibrofolliculomas, lung cysts, and the development of spontaneous pneumothorax.
Tuberous sclerosis, due to defects in TSC1 (located at 9q34) or TSC2 (16p13), may lead to clear cell RCC. More commonly, it is associated with multiple benign renal angiomyolipomas, renal cystic disease, cutaneous angiofibromas, and pulmonary lymphangiomyomatosis.
Succinate dehydrogenase RCC, due to defects in the SDHB (1p36.1-35) or SDHD (11q23) subunits of the succinate dyhydrogenase complex, may lead to a variety of RCC subtypes including chromophobe RCC, clear cell RCC, and type 2 papillary RCC. Extra-renal manifestations including benign and malignant paragangliomas and papillary thyroid carcinoma. In general, these tumors exhibit aggressive behaviour and wide surgical excision is recommended.
Finally, Cowden syndrome, due to defects in PTEN (10q23) may lead to papillary or other RCCs in addition to benign and malignant breast tumors and epithelial thyroid cancers.
Etiologic Risk Factors in Sporadic RCC
While numerous hereditary RCC syndromes exist, they account for only 4% of all RCCs. However, many sporadic RCCs share similar underlying genetic changes including VHL defects in ccRCC and c-Met activation in papillary RCC. A number of modifiable risk factors associated with RCC have been described.4
The foremost risk factor for the development of RCC is cigarette smoking. According to both the US Surgeon General and the International Agency for Research on Cancer, observational evidence is sufficient to conclude there is a causal relationship between tobacco smoking and RCC. A comprehensive meta-analysis of western populations demonstrated an overall relative risk for the development of RCC of 1.38 (95% confidence interval 1.27 to 1.50) for ever smokers compared to lifetime never smokers.7 Interestingly, this effect was larger for men (RR 1.54, 95% CI 1.42-1.68) than for women (RR 1.22, 95% CI 1.09-1.36). Additionally, there was a strong dose response relationship: compared to never smokers, men who smoked 1-9 cigarettes per day had a 1.6x risk, those who smoked 10-20 per days had a 1.83x risk, and those who smoked more than 21 per day had a 2.03x risk. A similar trend was seen among women. Notably, the risk of RCC declined with increasing durations of abstinence of smoking. Smoking appears to be preferentially associated with the development of clear cell and papillary RCC.8 In addition to being associated with increased RCC incidence, smoking is associated with more aggressive forms of RCC, manifest with higher pathological stage and an increased propensity for lymph node involvement and metastasis at presentation.9 As a result, smokers have worse cancer-specific and overall survival.9
Second, obesity is associated with an increased risk of RCC. While this risk was historically felt to be higher among women, a more recent review demonstrated no such effect modification according to sex.10 In a meta-analysis of 22 studies, Bergstrom et al. estimated that each unit increase of BMI was associated with a 7% increase in the relative risk of RCC diagnosis.
Third, hypertension has been associated with an increased risk of RCC diagnosis, with a hazard ratio of 1.70 (95%CI 1.30-2.22) in the VITAL study.11 Interestingly, in an American multiethnic cohort, this effect appeared to be larger among women (RR 1.58, 95% CI 1.09-2.28) than in men (RR 1.42, 95% CI 1.07-1.87).12 Again, as with obesity, there appears to be a dose-effect relationship between severity of hypertension and the risk of RCC diagnosis.13
Fourth, acquired cystic kidney disease (ACKD) appears to be associated with a nearly 50x increase risk of RCC diagnosis.14 ACKD occurs in patients with end-stage renal disease on dialysis. These changes are common among patients who have been on dialysis for at least 3 years.14 Interestingly, the risk of RCC appears to decrease following renal transplantation.
Finally, a number of other putative risk factors have been described. These lack the voracity of data that the aforementioned four have. Such risk factors include alcohol, analgesics, diabetes, and diet habits.4
Published Date: November 20th, 2018