Brief Overview of Adrenal Physiology
The adrenal is histologically divided into a three zoned cortex and the inner medulla. The adrenal cortex is involved in the multistep process of steroidogenesis. Each region of the cortex (glomerulosa, fasciculata, and reticularis) produces different steroidal end-products (mineralocorticoids, glucocorticoids, and androgens, respectively) as a result of differing ratios and types of enzymes that catalyze steroidogenesis. The adrenal medulla produces catecholamines (norepinephrine, epinephrine, and dopamine) under the control of the sympathetic branch of the autonomic nervous system.Adrenal Pathology
The differential diagnosis of an adrenal mass is broad, including a number of benign and malignant conditions as summarized in Table 1. In patients with bilateral adrenal masses, the differential diagnosis is somewhat shorter but includes metastases, congenital adrenal hyperplasia, adenomas, lymphoma, infectious causes, hemorrhage, pheochromocytoma, and amyloidosis, and ACTH-dependent Cushing's disease.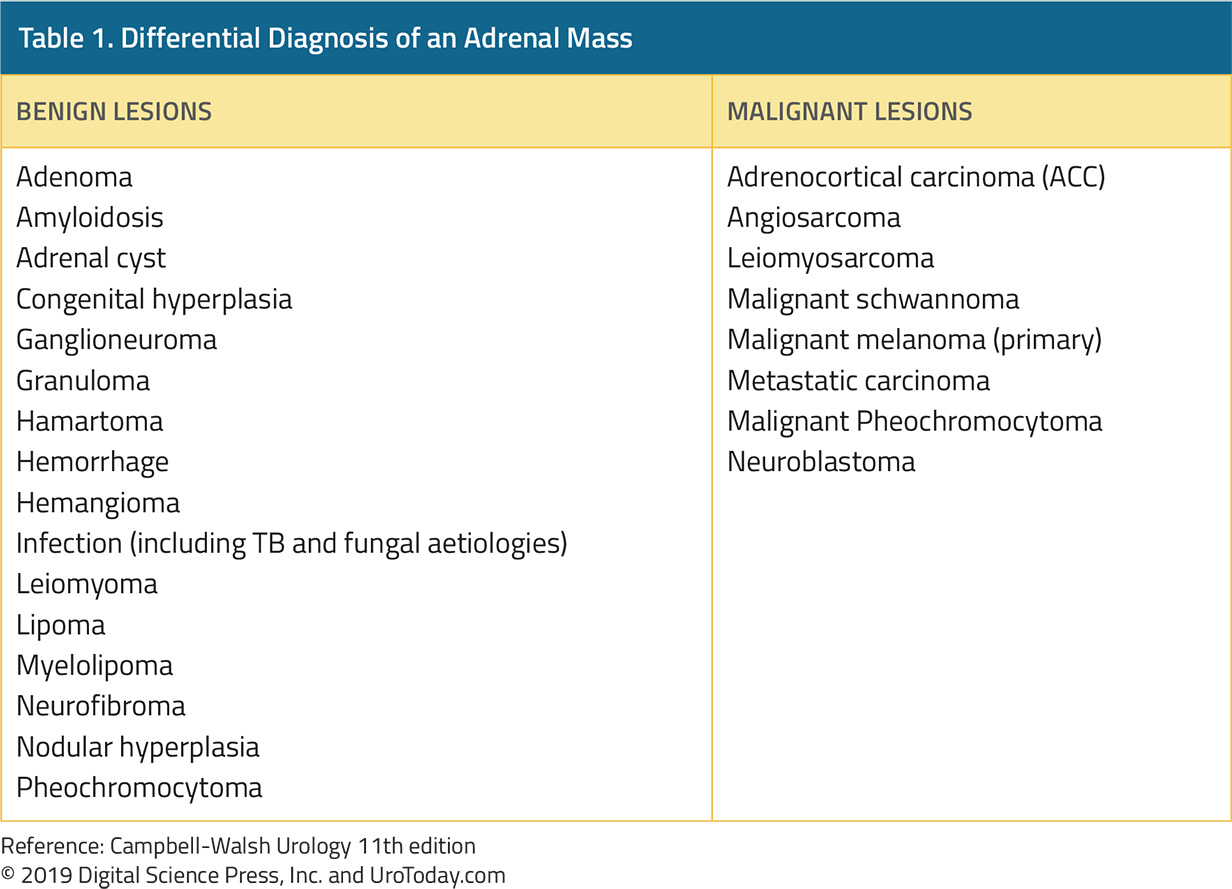
In urologic practice, many adrenal masses represent adrenal incidentalomas, masses >1 cm found on imaging performed for other reasons. While incidentally detected, a relatively large proportion (up to 20%) of these lesions may warrant surgical resection.1 Additionally, more than 10% of these lesions will prove to be biologically active. Therefore, metabolic testing (as detailed below) is recommended for all adrenal incidentalomas.2
Primary adrenal malignancies are uncommon. Adrenocortical carcinoma (ACC) has an incidence of less than 2 per million population.3 While there are associated hereditary syndromes, the majority of ACCs are sporadic. ACC may be biochemically functional or non-functional. Among functional lesions, hypercortisolism is the most common.
From an oncologic perspective, metastases are a much more common cause of adrenal lesions than primary adrenal pathology. Primary cancers with a particular predilection for adrenal metastases including melanoma, lung cancer, renal cell carcinoma, breast cancer, and medullary thyroid cancer.4 However, a wide variety of other cancer may also spread to the adrenal. In patients with known extra-adrenal malignancy, a new adrenal mass is likely to represent metastasis in approximately 50% of cases4. Thus, standard functional assessment is advocated.4
While we will not dwell on it further, a brief mention of congenital adrenal hyperplasia is warranted. This is an autosomal recessive congenital condition characterized by low cortisol production as a result of enzymatic defects in the steroidogenesis pathway. Deficiency in 21-hydroxylase is the cause of nearly 95% of cases. Due to a lack of feedback, there is overproduction of ACTH and resulting overproduction of adrenal androgens. This condition is most often diagnosed and managed in childhood, thus, it will be uncommon as a presentation for adults with newly diagnosed adrenal lesions.
Investigation of Adrenal Lesion
With a newly identified adrenal lesion, there are two primary questions which will guide further management. First, could this mass be malignant? Second, is this mass functional? That is, are there any physical signs and symptoms or biochemical evidence of excess hormonal activity that could be attributed to excess secretion of an adrenally derived hormone.Imaging is warranted (and likely the reason for assessment) for patients with adrenal lesions. Ultrasound is relatively poor at visualizing and characterizing adrenal lesions. Therefore, axial imaging using CT or MRI is advised. Unenhanced CT scan is the first line test of choice. In more than 70% of cases, it is possible to identify adrenal adenomas on the basis of this test alone. Low attenuation (<10 HU) is the characteristic finding on this study. Enhanced CT with adrenal washout protocols may be used where unenhanced CT is unclear. Adenomas exhibit characteristic rapid enhancement washout after administration of CT contrast. MRI is an alternative to CT scan. Again, there are characteristic findings of adrenal adenomas including a loss of signal intensity of out-of-phase sequences.5
Imaging findings help to guide the answer to the question of whether a given adrenal lesion may be malignant. There is a relationship between the size of an adrenal lesion and the likelihood of malignancy. Thus, all lesions larger than 6 cm should be considered malignant until proven otherwise. Due to diagnostic uncertainty, may would advocate resection for lesions 4 cm or larger.1 Additionally, as the incidence of benign lesions increases with age, additional concern should be taken for younger patients with even small adrenal lesions. On axial imaging, ACC exhibit increase attenuation on non-contrast CT, irregular borders and enhancement, and calcification and necrosis.
Functional assessment of adrenal lesions begins with history and physical examination. Cushing's syndrome, caused by excess production of glucocorticoids, may present with central obesity, proximal muscle weakness, thinning of the skin, a so-called buffalo hump, or moon facies. Primary hyperaldosteronism, also known as Conn’s disease, may present with hypertension and hypokalemia. In many patients, hypertension is quite severe with mean blood pressures in the range of 180/1106. Pheochromocytomas, which secrete catecholamines, may present with hypertension, arrhythmia, anxiety, headache, pallor, diaphoresis, and tremor. The classic triad comprised headache, episodic sudden perspiration and tachycardia.7 Adrenocortical carcinoma may produce functional syndromes as described above or may also cause mass-related effects including abdominal fullness, back pain, nausea, and vomiting.
Biochemical assays are employed to confirm functional lesions. For Cushing's syndrome, the diagnosis may be confirmed with a 24-hour urinary free cortisol test or a low-dose dexamethasone suppression test. Following diagnosis, a number of subsequent tests may be performed to ascertain the underlying etiology. While these are typically coordinated by an endocrinologist, they will be briefly summarized here. Determination of serum ACTH (adrenocorticotropic hormone) can distinguish ACTH-dependent Cushing’s from ACTH-independent causes. Among patients with elevated ACTH, determination of the anatomic source, whether pituitary or ectopic, can drive further management. However, modern imaging remains relatively insensitive and non-specific for the detection of both pituitary and ectopic sources of ACTH.8,9 Therefore, direct measurement of venous levels of ACTH in the inferior petrosal sinus following CRH stimulation has been accepted to distinguish pituitary and ectopic sources of ACTH.8 High-dose dexamethasone suppression testing is no longer routinely used.8
Due to the underlying pathophysiology, patients must stop mineralocorticoid receptor antagonist antihypertensives prior to investigation for primary hyperaldosteronism. Further, hypokalemia should be corrected. For these patients, it is critical to determine whether this is a primary process or driven by perturbations in renin levels. Thus, determination of the ratio of serum aldosterone to plasma renin activity (PRA) is critical. This is known as the aldosterone to renin ratio (ARR). For patients with a positive ARR screening test, confirmatory testing typically seeks to identify suppression of aldosterone production following sodium loading. Options include fludrocortisone suppression testing, oral sodium loading, and intravenous saline infusion. Other, less commonly utilized, tests include captopril suppression testing, the furosemide-upright test, and the ACTH stimulation test. However, a number of etiologies may contribute to primary hyperaldosteronism including bilateral or unilateral hyperplasia, adenomas, and tumors. Therefore, following confirmation, subtype investigations may be undertaken among patients who are surgical candidates. This is typically performed with cross-sectional imaging. For patients without identified unilateral nodules, adrenal venous sampling may allow lateralization of the lesion. In the case of a non-diagnostic sampling, other optics including nuclear scintigraphy and postural stimulation testing.
Pheochromocytomas are potentially the most worrisome of functional adrenal lesions given the potential for significant cardiovascular instability if they are not recognized prior to intervention. Evaluation of these masses should include both biochemical and radiographic studies. Biochemical studies assess catecholamines and their metabolites including plasma free metanephrines, catecholamines, urinary fractionated metanephrines, total metanephrines, and vanillylmandelic acid. Each of these tests have varying sensitivity and specificity. Today, most advocate testing of plasma free metanephrine levels10 as this is more sensitive than serum levels of catecholamines. For patients with equivocal findings, use of the clonidine suppression test has been suggested by some.11 Chromogranin A is an alternative confirmatory test though the sensitivity is somewhat poor for this function.
As with all adrenal lesions, imaging of pheochromocytoma begins with computed tomography (CT). Unlike adrenal adenomas, pheochromocytoma typically has an increased attenuation (mean 35 HU).12 Magnetic resonance imaging (MRI) is an alternative. Classically, these lesions have a bright signal, termed the "light bulb" sign. Functional imaging may be undertaken using 18F-FDG PET scanning or metaiodobenzylguanidine (MIBG) scintigraphy.
As hereditary lesions account for nearly 1/3 cases of pheochromocytoma, familial testing has been suggested among patients who have a family history, present at age <50 years, have multiple lesions, malignant pheochromocytoma, or bilateral pheochromocytoma.13
Investigations to assess the functionality of adrenal lesions are summarized in Table 2.

Investigation of suspected ACC should assess excesses of glucocorticoids, sex steroids, catecholamines, and mineralocorticoids. The Weiss pathologic criteria are used to distinguish benign and malignant adrenal lesions (Table 3).14 The presence of three or more of these criteria is highly associated with malignancy.

Treatment of Adrenal Lesions
For patients with small non-functional adrenal lesions with benign imaging findings, surveillance may be appropriate. However, surgery is the mainstay for patients with adrenal lesions. There are particular nuances on the basis of the underlying histology and functional status. In general, laparoscopic adrenalectomy is considered the gold standard as, in experienced hands, oncologic outcomes are equivalent with improved convalescence.For patients with adrenocortical carcinoma, surgical resection is the standard of care. In these cases, wide margins are critical. Thus, for larger tumors with possible adjacent organ involvement, some authors advocate that these cases should be performed open in order to ensure negative margins given the potential need for adjacent organ resection. Unfortunately, recurrence is common following even aggressive resection. Radiotherapy can be used in an adjuvant setting for patients with positive margins and for treatment of bone or central nervous system metastases. Systemic therapy may be undertaken with mitotane, a synthetic derivative of DDT.
For patients with Cushing's disease, the management varies widely based on underlying etiology. The overall goals included correction of the cortisol excess, restoration of the underlying hormonal axis, and management of the sequelae. Approaches to this, depending on underlying etiology, include weaning of exogenous steroids, transsphenoidal resection of pituitary lesions, unilateral or bilateral adrenalectomy, resection of ectopic sources of ACTH, and medical therapy with blockers of steroidogenesis.
Treatment of primary aldosteronism seeks to control blood pressure and prevent sequelae of hormonal excess. This may be accomplished medically or surgically depending on the underlying cause and patient suitability for operation. Medical treatment may be undertaken with aldosterone receptor antagonists such as spironolactone or eplerenone.
Pheochromocytoma is primarily a surgical disease. However, extensive medical consultation and optimization is required to prevent significant intraoperative cardiovascular complications. Further, these patients are at risk of cardiomyopathy and, therefore, consultation with a cardiologist or anesthesiologist prior to surgery is advisable. Catecholamine blockade is required prior to surgery on pheochromocytoma. Classically, this has been achieved with the non-competitive alpha-blocker phenoxybenzamine. However, selective reversible alpha-blockers including doxazosin or terazosin are alternatives. Following alpha-blockade, beta-blockade may be undertaken due to the risk of reflex tachycardia or arrhythmia.13 An alternative to alpha- and beta-blockade which has been proposed utilized calcium channel blockade.15 Finally, catecholamine synthesis blockade through the use of alpha-methyltyrosine (metyrosine) may be added. In the perioperative period, repletion of the intravascular volume is critical. This may be achieved through liberal consumption of salt and liquid or intravenous resuscitation. Careful postoperative monitoring is key as these patients are at risk for hypotension and hypoglycemia. Additionally, as these lesions have a predilection for recurrence, ongoing monitoring is required.
Published Date: January 28th, 2019