Background
Kidney cancer is the 12th most common cancer in the world, with over 300,000 new cases annually, of which 65,340 new cases will be diagnosed in the United States in 2018.1 The incidence of renal cell carcinoma (RCC) varies substantially based on the country – rates of RCC are higher in Europe and North America and much lower in Asia and South America.2
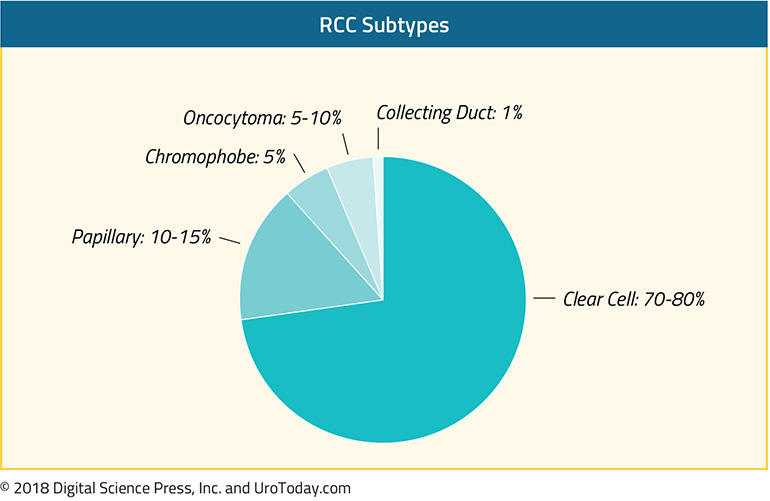
Most kidney cancers (>90%) are renal cell carcinomas, and of renal cell carcinomas, the majority of cases (80%) will be the clear cell subtype.3 Of the remaining 20% of cases, the two major histological subtypes are papillary (10-14%) and chromophobe (5%)3,4, but also include collecting duct, translocation carcinoma, medullary carcinoma, and unclassified RCC. These histological subtypes are distinct from clear cell carcinoma and independently predict for survival.3 For example, after controlling for TNM status, age, gender, and tumor size, patients with early stage clear cell RCC are more than twice as likely to die of RCC than patients with papillary or chromophobe RCC.3 Some of these subtypes also have unique risk factors. For example, renal medullary carcinomas are an aggressive non-clear cell carcinoma that are almost exclusively associated with patients with sickle cell trait.5
Non-clear cell RCCs (ncRCC) also have unique mutational landscapes.6 For example, MET mutations can be found in 15-30% of papillary RCCs.6,7 Papillary RCCs also have a higher mutation rate compared with chromophobe RCCs and renal oncocytomas. Durinck et al evaluated 167 primary human tumors including papillary, chromophobe and translocation subtypes and were able to use a five gene set to help molecularly distinguish between chromophobe carcinoma, renal oncocytoma and papillary carcinoma.6
Treatment
Given the rarity of non-clear cell RCC (ncRCC), there is a paucity of large randomized phase III trials to help guide the optimal therapy for ncRCC. Some trials for ncRCC have had to stop due to slow accrual. Given this lack of data, patients are encouraged to participate in clinical trials when they are available and appropriate. Below is a summary of the most common systemic therapies in use for ncRCC.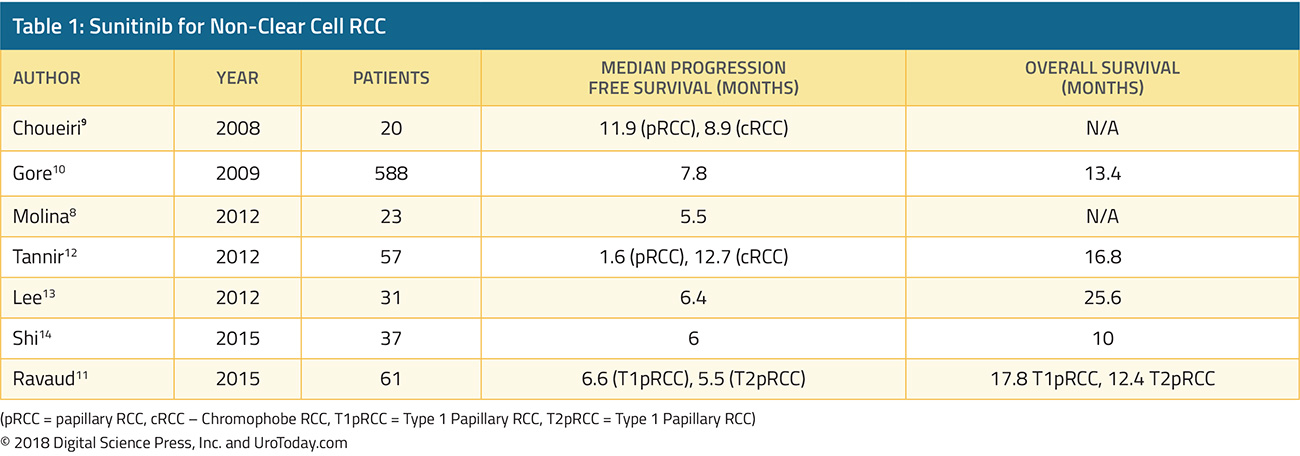
Sunitinib
Single agent sunitinib has been evaluated as part of an expanded access trial as well as several small phase II trials (Table 1). In a single arm phase II clinical trial, 23 patients were given sunitinib 50 mg in cycles of four weeks on followed by 2 weeks off. The trial was stopped early due to slow accrual and the median progression free survival (PFS) was 5.5 months.8 In a another phase II trial evaluating both sunitinib and sorafenib, 19 patients with ncRCC were given sunitinib – median PFS was 11.9 months in the papillary arm and 8.9 months in the chromophobe arm.9 The largest of these studies was reported by Gore et al, which included 588 ncRCC patients who received sunitinib as part of the expanded access study encompassing 4564 RCC patients who received sunitinib.10 In their study, 11% of patients had an objective response and median PFS was 7.8 months. Some trials have stratified outcomes by histological subtypes such as chromophobe or papillary, and one trial reported results broken down by type 1 papillary RCC vs type 2 papillary RCC11. Overall, most studies demonstrated that sunitinib led to a median PFS of about 6-7 months for ncRCC.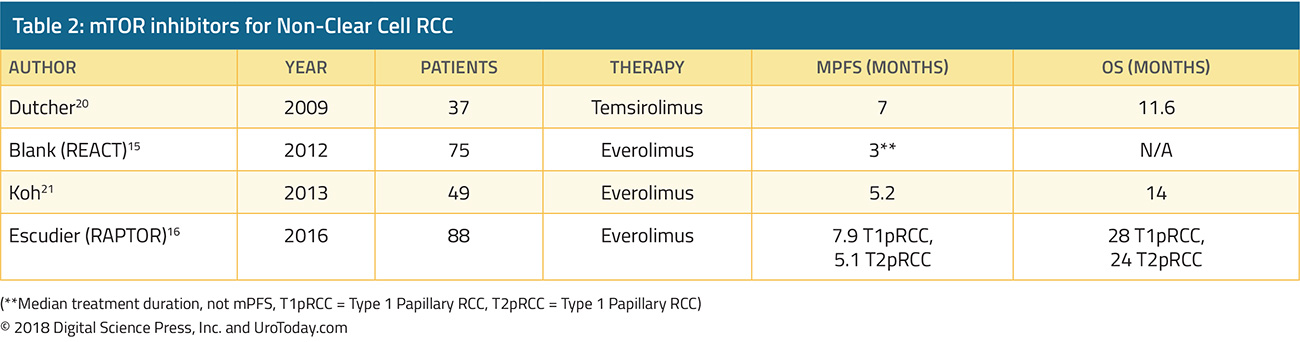
Everolimus and Temsirolimus
mTOR inhibitors Everolimus and Temsirolimus have been evaluated by a few phase II trials for ncRCC (Table 2). Everolimus was evaluated in a subgroup of REACT (RAD001 Expanded Access Clinical Trial in RCC) and in RAPTOR (RAD001 in Advanced Papillary Tumor Program in Europe).15,16 In REACT, 1.3% of patients with ncRCC had a partial response and 49.3% had stable disease.15 In RAPTOR, the median progression free survival (mPFS) was 4.1 months and median OS was 21.4 months. For patients with chromophobe RCC, mTOR directed therapy may be especially effective – case reports show partial responses and stable disease to both Everolimus or Temsirolimus.17,18 This may be due to the hypothesis that a high number of chromophobe RCC’s have PI3K-mTOR pathway activation as well as frequent TSC1/TSC2 mutations which may sensitize these tumors to mTOR inhibition.19
Sunitinib vs Everolimus
Sunitinib and everolimus have also been examined head to head. The largest trial was ASPEN (Everolimus versus sunitinib for patients with metastatic non-clear-cell renal cell carcinoma), a multicenter open-label, randomized phase II trial which randomized 108 patients to receive either sunitinib or everolimus.22 The primary endpoint of this study was progression free survival. The majority of patients had papillary histology (65%). Median overall survival was 13.2 months in the everolimus group and 31.5 months in the sunitinib group. However, overall survival was not statistically different between the two treatment groups (HR 1.12, 95%CI 0.7-2.1, p=0.6). A second study, ESPN (Everolimus Versus Sunitinib Prospective Evaluation in Metastatic Non–Clear Cell Renal Cell Carcinoma) came to a similar conclusion and found that the median overall survival was 16.2 months with sunitinib and 14.9 months with everolimus (p=0.18).23
While overall survival between everolimus and sunitinib were not statistically different for the unselected cohorts, ASPEN did find differences in objective responses between the different subtypes, suggesting that each subtype of ncRCC may respond differently to therapies. In ASPEN, 24% (8/33) of papillary RCCs achieved a partial or complete radiographic response on sunitinib compared with 5% of patients on everolimus (2/37). Interestingly, clinical outcomes after receipt of either sunitinib or everolimus also varied based on risk stratification. Patients with good or intermediate risk had improved median progression free survival (mPFS) with first line sunitinib than everolimus. However, patients with poor risk had improved PFS with everolimus over sunitinib. This is concurrent with the ARCC trial, which also demonstrated improved overall survival with an mTOR inhibitor (temsirolimus) in poor risk patients with ncRCC.24
A meta-analysis of five studies (ESPN, ASPEN, RECORD3, ARCC, and SWOG1107) found a nonstatistical trend favoring sunitinib over everolimus for ncRCC but does note that there is considerable patient heterogeneity in these small studies and there was no statistical difference in PFS between these two therapies.25 In the absence of clinical trial options, sunitinib is a reasonable first line choice for treatment naïve patients with ncRCC, especially for those with papillary RCC or MSKCC good risk RCC.
Special Populations
It is well recognized that ncRCC is a heterogenous mix of patients which respond differently to therapies. Thus, there have been a few biomarker or histology driven trials looking at specific subsets of ncRCC. For example, for patients with hereditary leiomyomatosis and papillary RCC, a phase II study of 41 patients found that patients treated bevacizumab plus erlotinib had a median PFS of 24.2 months, compared to 7.4 months for patients with sporadic papillary RCC.26 Patients with papillary RCC frequently have MET mutations and a variety of MET inhibitors including crizotinib, savolitinib, and cabozantinib are being evaluated in clinical trials.27-29 For example, in a study of 41 patients with type 1 papillary RCC, of the 4 patients with MET+ tumors, 2 had achieved a partial response and one had stable disease with crizotinib.28 In a study with Savolitinib, another selective MET inhibitor, patients with MET “driven” tumors had a median PFS of 6.2 months, compared with 1.7 months for MET independent tumors.29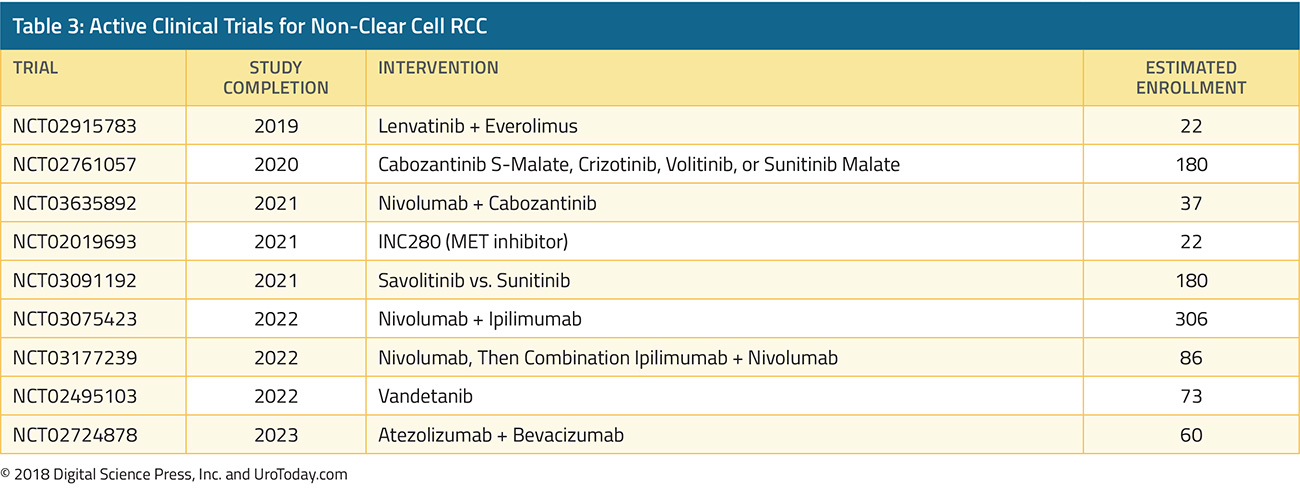
Future Direction
A number of active clinical trials are in progress, investigating various MET inhibitors as well as checkpoint inhibitors for ncRCC (Table 3). Preliminary data suggest that PD-1 or PD-L1 blockade may have some activity in this population.30,31 Given the dearth of data and rarity of ncRCC, it is important to consider these patients for clinical trials, whenever possible.
Published Date: November 29th, 2018