Highlights
1. Poly (ADP-ribose) polymerases (PARPs) play a key role in several DNA-damage response (DDR) pathways2. PARP inhibition in cells with defective double-strand DNA-repair (e.g. due to BRCA1 or BRCA2 mutation) results in synthetic lethality
3. Two PARP inhibitors (rucaparib and olaparib) have been recently FDA-approved for the treatment of metastatic castration-resistant prostate cancer harboring selected homologous recombination repair (HRR) gene mutations and progressing on at least one novel antiandrogen therapy and a chemotherapy
4. PARP monotherapy has a tolerable side effect profile and combination approaches such as rucaparib with enzalutamide are in various phases of development
Background
First studies of PARP inhibitors such as nicotinamide date back to the 1970s and initial development was focused on using these drugs as radio- or chemo-sensitizers.1,2 Two seminal studies showed that in the absence of adequate homologous recombination (HR) repair, tumor cells are exquisitely sensitive to PARP inhibition due to PARPs' complementary roles in single-strand break (SSB) and non-homologous end joining (NHEJ) repair pathways.3,4 This pre-clinical demonstration prompted a slew of clinical trials that unequivocally established PARP inhibitors as a new targeted therapy for multiple tumor types harboring aberrant HR repair pathways.Overview of FDA-approved indications for PARP inhibitors
Four PARP inhibitors (rucaparib, olaparib, niraparib, and talazoparib) have been approved by the US Food and Drugs Administration (FDA) for treatment of human malignancies as of June 2020.5 Olaparib is the only PARP inhibitor that has been FDA-approved in combination with another agent.5Table 1. Summary of PARP inhibitor approvals
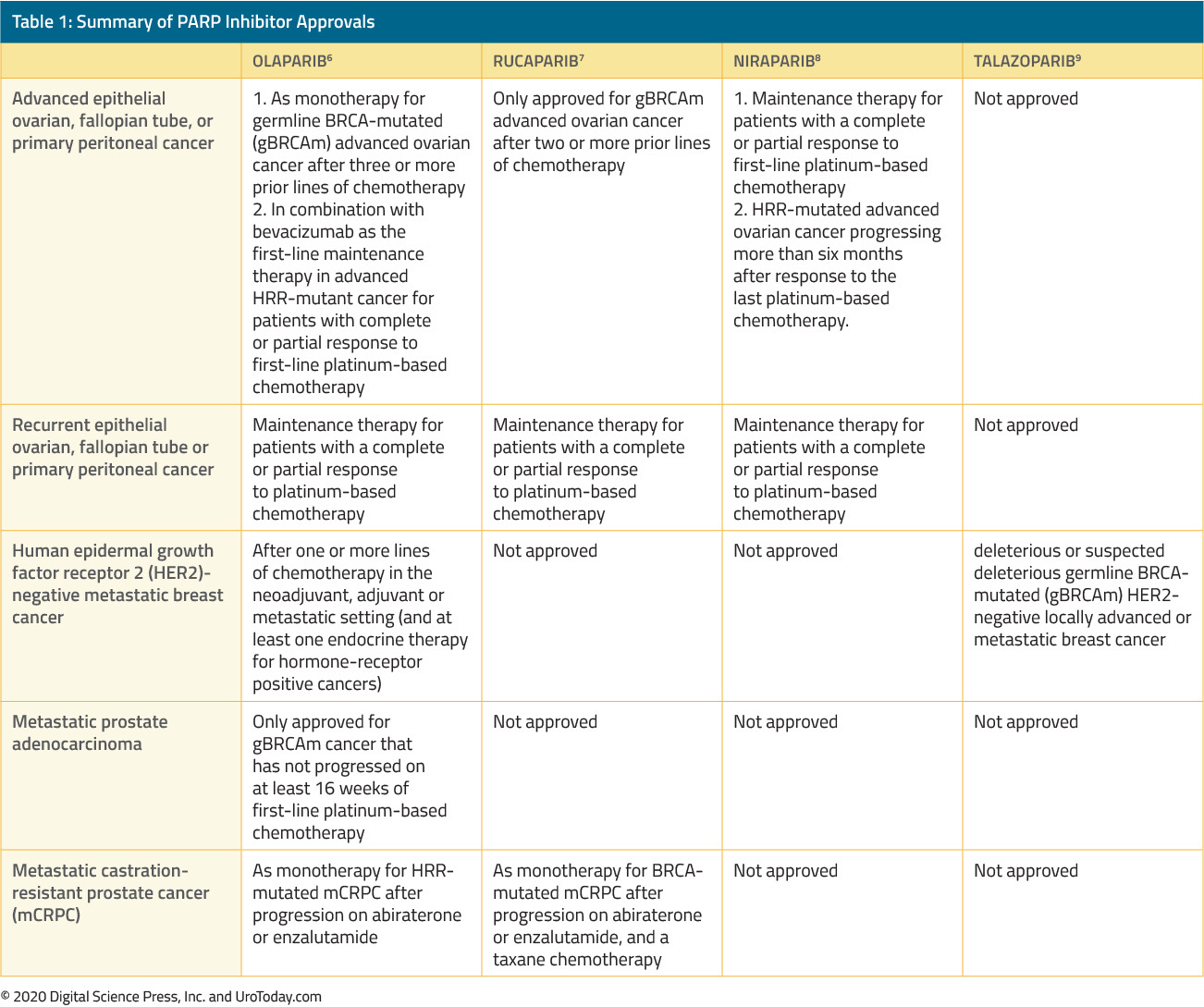
PARP inhibitor monotherapy in mCRPC
Rucaparib, and subsequently, olaparib, were FDA-approved in May 2020 for use in metastatic castration-resistant prostate cancer (mCRPC) harboring certain HRR aberrations.6,7 Preliminary results of the trials evaluating niraparib and talazoparib have also shown encouraging activity. The data on the efficacy of these agents appears to be comparable and is detailed below.Olaparib
TOPARP-A was a pivotal Phase II trial of olaparib in mCRPC in which 50 patients were treated with olaparib 400 mg twice daily until disease progression.10 The primary endpoint was the composite response rate defined either as an objective response according to Response Evaluation Criteria in Solid Tumors (RECIST) criteria, or a ≥ 50% reduction in prostate-specific antigen (PSA), or a reduction in the circulating tumor-cell count from ≥ 5 per 7.5 ml of blood to < 5 per 7.5 ml. All patients had prior treatment with docetaxel and 49 (98%) with abiraterone or enzalutamide. Sixteen of 49 (33%) evaluable patients had a response. Remarkably, however, 14 of the 16 responders had homozygous deletions, deleterious mutations, or both in DNA-repair genes — including BRCA1/2, ATM, Fanconi’s anemia genes, and CHEK2.
This was followed by TOPARP-B, an open-label, Phase II trial in which men with HRR-mutated mCRPC that had progressed on at least one taxane therapy were treated with olaparib 400mg or 300mg twice daily in a randomized fashion.11 The primary endpoint was identical to the TOPAPRP-A trial. A targetable HRR gene aberration was found in 161 of 592 (27.2%) patients who underwent a targeted next-generation tumor sequencing. However, sequencing could not be performed on 119 (17%) of consented patients because of insufficient or poor quality tissue. The confirmed composite response rate was 54.3% (95% confidence interval [CI] 39.0–69.1) in the 400 mg cohort and 39.1% (95% CI 25.1–54.6) in the 300 mg cohort (p=0.14). Median radiographic progression-free survival (rPFS) was 5.5 months (95% CI 4.4–8.3) in the 400 mg cohort and 5.6 months (3.7–7.7) in the 300 mg cohort. The predefined criteria for success were met for the 400 mg regimen but not for the 300 mg regimen.
PROfound was a randomized, open-label, Phase III trial of olaparib 300mg twice daily versus physician’s choice of standard-of-care (SOC) therapy in men with HRR-mutated mCRPC who had disease progression while receiving a novel hormonal agent (e.g., enzalutamide or abiraterone)12. Patients were assigned to one of two cohorts based on the HRR gene alteration. Cohort A had patients with BRCA1, BRCA2, or ATM alteration regardless of co-occurring alteration in any other HRR gene. Cohort B had patients with alterations in any of the other 12 HRR genes. Patients within each cohort were randomized in 2:1 fashion to olaparib versus SOC. The primary endpoint was the rPFS in cohort A.
Of the 4425 enrolled patients, 4047 had tumor tissue available for testing and only 2792 (69%) were successfully sequenced. A qualifying alteration in one or more of the 15 HRR genes was detected in 778 of 2792 patients (28%). Median rPFS was significantly longer in the olaparib group than in the SOC group (7.4 months vs. 3.6 months; hazard ratio [HR] 0.34; 95% CI, 0.25 to 0.47; P<0.001). The confirmed objective response rate was 33% in the olaparib group and 2% in the SOC group (odds ratio 20.86; 95% CI, 4.18 to 379.18; P<0.001). The median time to pain progression was also significantly longer in the olaparib group (HR, 0.44; 95% CI, 0.22 to 0.91; P=0.02). An interim analysis for overall survival (OS) at 38% data maturity showed that median OS was longer in the olaparib group than in the SOC group (18.5 months vs 15.1 months; HR 0.64; 95% CI, 0.43 to 0.97; P=0.02). Of note, 81% of patients in the SOC group had crossed over to receive olaparib treatment at the time of this analysis. Thus, olaparib is unequivocally more effective than SOC therapy in this patient population.
The data from PROfound trial formed the basis for FDA-approval of olaparib 300mg PO twice daily in men with HRR-mutated mCRPC after progression on enzalutamide or abiraterone.
Rucaparib
TRITON2 is an ongoing, multi-center, single-arm clinical trial of rucaparib in men with HRR-mutated (germline and/or somatic) mCRPC who have previously received at least one novel antihormonal agent and taxane-based chemotherapy. As of Feb 2019, 190 patients had received rucaparib 600 mg orally twice daily13. Confirmed overall response rate (ORR) was 43.9% (25/57; 95% CI 30.7%–57.6%) in patients with a BRCA1/2 alteration and RECIST-defined measurable disease at baseline. ORR was comparable in patients with a germline BRCA1/2 alteration (38.1%; 95% CI, 18.1%–61.6%) and somatic BRCA1/2 alteration (48.6%; 95% CI, 31.4%–66.0%). The median duration of response was not reached but the range was 1.7 to 24+ months.
The data from the TRITON2 trial formed the basis for this accelerated approval of rucaparib 600mg PO twice daily in men with HRR-mutated mCRPC after progression on enzalutamide or abiraterone and a taxane-based chemotherapy7.
A confirmatory trial TRITON3 is currently ongoing. The design of this trial is similar to the PROfound trial as above, with 400 men with BRCA1/2 or ATM-mutated mCRPC randomized to rucaparib or SOC therapy in 2:1 fashion. The primary endpoint is rPFS and the results are expected by 2022.
Niraparib
GALAHAD is an ongoing, open-label, Phase II trial of niraparib in men with HRR-aberrant mCRPC that has progressed after at least one novel antihormonal agent and taxane-based chemotherapy. The primary endpoint is ORR by RECIST 1.1 with no evidence of bone progression (per PCWG3 criteria). Composite ORR, similar to that used in TOPARP-B and TRITON2 trials, is also evaluated. As of May 2019, 223 patients were screened and 165 were enrolled, of whom 81 (46 BRCA1/2 and 35 non-BRCA) had biallelic HRR aberrations and were treated with niraparib 300mg daily14. In BRCA1/2-aberrant patients, ORR was 41% and composite ORR was 63%. Median rPFS for BRCA-aberrant patients was 8.2 months and median OS was 12.6 months. In non-BRCA-aberrant patients, ORR was 9% and composite ORR was 17%. The median rPFS for non-BRCA-aberrant patients was 5.3 months and OS was 14.0 months.
Talazoparib
TALAPRO-1 (NCT03148795) is an ongoing Phase II trial of talazoparib in men with HRR-aberrant mCRPC that has progressed after at least one novel antihormonal agent and taxane-based chemotherapy. The primary endpoint is ORR by RECIST 1.1. Data from 81 of the planned 100 patients were reported recently.15 In the 43 evaluable patients who received talazoparib 1mg daily, ORR was 25.6% (13.5–41.2). ORR in BRCA1/2-aberrant population was 50.0% (27.2–72.8) and ORR in ATM-aberrant population was only 7.1% (0.2–33.9). Overall median (95% CI) rPFS was 5.6 months (3.5–8.2), and that in BRCA1/2-aberrant and ATM-aberrant populations is 8.2 mo (5.6–NE) and 3.5 mo (1.7–8.1), respectively.
Management of side effects of PARP inhibitors
Adverse event profiles of all PARP inhibitors overlap considerably. The most common (any CTCAE grade) clinical side effects in Phase III trials of rucaparib, olaparib and niraparib were fatigue (60-70%), nausea (~75%), vomiting (~35%), dysgeusia (10-40%), anorexia (~25%), diarrhea (20-30%), constipation (20-40%), abdominal pain (25-30%), headache (20-25%) and cough (10-15%). The most common (any CTCAE grade) lab abnormalities were anemia (40-50%), thrombocytopenia (15-60%), neutropenia (20-30%), increase of alanine aminotransferase (ALT; 5-36%), increase of aspartate aminotransferase (AST; 2-28%), and increased serum creatinine level (10-15%).16While nausea is the most common side effect of PARP inhibitor therapy, it tends to be mild in most cases. We routinely recommend taking the medication after a meal and an antiemetic (prochlorperazine or a 5-HT3 antagonist such as ondansetron) in patients who develop moderate or severe nausea and/or vomiting with PARP inhibitor therapy.
Thrombocytopenia appears to be more common with niraparib at 61%, as opposed to olaparib (14%) or rucaparib (28%). The niraparib FDA label thus recommends obtaining weekly platelet levels during the first month of therapy.8 Elevated serum creatinine level occurs within the first few weeks of therapy and is thought to be an on-target effect due to the inhibition of renal transporter proteins. Thus, serum creatinine-based estimation of renal function may not be accurate in patients receiving PARP inhibitor therapy. Alternative methods of glomerular filtration rate (GFR) estimation such as radionuclide scan or serum-cystatin C must be used in cases where a more accurate GFR estimate is necessary. Elevation of AST and ALT also tends to typically occur within the first two cycles and can be transient. Treatment interruption may not be required for mild AST/ALT elevations but serum bilirubin levels must be checked in all patients to evaluate for drug-induced liver injury.7
Owing to their mechanism of action, there was a concern regarding treatment-emergent myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) with PARP inhibitor therapies. However, the current experience puts the risk of MDS/AML at under 1.5%. Of the 2351 patients treated in olaparib monotherapy trials, only 28 (<1.5%) developed MDS/AML. Of these, 25/28 patients had a BRCA mutation, two patients had a wild-type gBRCA and one had unknown BRCA mutation status. The duration of olaparib varied from < 6 months to > 2 years and all had received previous chemotherapy with platinum and/or other DNA damaging agents, or radiotherapy. If pancytopenia occurs at any point during PARP inhibitor therapy, treatment must be interrupted as per guidelines for the drug, and appropriate evaluation for MDS and AML must be undertaken. Therapy must be discontinued permanently if a diagnosis of MDS or AML is confirmed.6
Another important consideration is the potential for often clinically-significant drug-drug interactions (DDI) with all PARP inhibitors. Rucaparib and olaparib are primarily metabolized by different members of the cytochrome P450 enzyme family, resulting in only a partial overlap in DDI16. Niraparib is metabolized in the liver by carboxylesterase-catalyzed amide hydrolysis with cytochrome P450 playing only a negligible role.17(p20) Many commonly-used drugs (such as phenytoin, carbamazepine, ketoconazole, ciprofloxacin, digoxin) have uni- or bi-directional interactions with PARP inhibitors. Thus, careful attention must be paid to minimize DDI by avoiding, discontinuing, adjusting the dose, or clinical/lab monitoring of these medications before and during PARP therapy. A nuanced discussion of these interactions is outside the scope of this article, but we routinely request a DDI review by a dedicated oncology pharmacist in our clinical practice.
How to choose PARP inhibitor for prostate cancer patients
Currently, the use of any of the PARP inhibitors as monotherapy in mCRPC requires the presence of deleterious alterations in the HRR gene as identified on a commercial assay (such as those resulting in protein truncation - e.g. nonsense, frameshift, or consensus splice site alterations; select missense alterations that are well-known to be deleterious; or homozygous deletions). Patients must have received at least one line of prior therapy with abiraterone or enzalutamide (olaparib and rucaparib), and one line of therapy with taxane chemotherapy (rucaparib). The completion of ongoing studies with rucaparib, niraparib, and olaparib may broaden the clinical states in which they are indicated.No dose modifications are required for olaparib, rucaparib or niraparib in patients with mild or moderate hepatic impairment, and for talazoparib in patients with mild hepatic impairment. Olaparib and talazoparib require dose reduction in patients with moderate (GFR 30-59 ml/min) or severe (GFR 15-29) renal impairment.
There are differences in side effect profiles of PARP inhibitors as detailed above, which result in differences in monitoring while on therapy. For example, the niraparib FDA-label recommends weekly complete blood count, blood pressure, and heart rate checks for the first month.8
Going beyond PARP inhibitors - Directions for the future
Despite excellent activity, only 15-25% of mCRPC patients will be eligible for PARP inhibitor monotherapy and nearly all patients treated with one will progress within a year.18 Several approaches are under investigation to overcome these issues. To date, the only PARP-based combination therapy that has been FDA-approved is olaparib plus bevacizumab in advanced ovarian cancer as mentioned above. Trials evaluating a combination of PARP and chemotherapy and immunotherapy are ongoing (Table 2).Induction of synthetic lethality using co-inhibition of androgen receptor (AR) signaling and PARP have become an area of intense research activity. Substantial pre-clinical evidence suggests that AR signaling is important in double-strand break (DSB) repair and its interruption results in genomic instability in non-HRR-aberrant prostate cancer models.19,20 This provides an effective mechanism of synergy with PARP inhibition. A recent trial of olaparib and abiraterone demonstrated that this phenomenon may be occurring clinically as well.21 A randomized, placebo-controlled Phase III trial of rucaparib and enzalutamide in untreated mCRPC (A031902; CASPAR) is expected to launch soon and several others are currently ongoing (PROpel [NCT03732820], MAGNITUDE [NCT03748641], TALAPRO-2 [NCT03395197]).
Table 2. A list of selected PARP inhibitor combination therapy trials

Written by: Arpit Rao, MBBS, Assistant Professor of Medicine, Division of Hematology, Oncology and Transplantation, and Charles Ryan, MD, Professor of Medicine, Division of Hematology, Oncology and Transplantation, Director, Division of Hematology, Oncology and Transplantation, B.J. Kennedy Chair in Clinical Medical Oncology, University of Minnesota Medical School, Minneapolis, Minnesota
Published June 2020
Related Content:
- PARP Inhibitors, Prostate Cancer and a Promise Fulfilled
- Germline Testing for DNA Repair Mutations in Prostate Cancer: Who, When and How?
- What Are the Most Common Genomic Aberrations Seen in DNA Damage Response (DDR) Pathways in Advanced Prostate Cancer?
- DNA Repair and PARP Inhibitor Therapy in Prostate Cancer - Veda Giri, Patrick Pilié, and Arpit Rao