What are the most common genomic aberrations seen in DNA damage response (DDR) pathways in advanced prostate cancer?
Men with advanced prostate cancer have a 10-15% risk of carrying a hereditary, or germline, variant in a DNA damage response (DDR) gene, as previously discussed. Pathogenic or deleterious variants in these same DDR genes can also be found at the somatic, or tumor-associated level, in up to 25% of metastatic castrate-resistant prostate cancer.1 Precision medicine currently centers mostly on discovering these somatic aberrations through DNA sequencing to then guide targeted treatment selection for patients with advanced cancer. Compared to other solid tumors such as melanoma or urothelial cancers, advanced prostate cancer overall displays relatively low tumor mutational burden (TMB), with rare exceptions including those tumors with mismatch repair (MMR) deficiency and/or subsequent high microsatellite instability (MSI-H). Defective MMR genes and/or MSI-H are seen in ~3-8% of prostate cancer, with the majority being of sporadic origin and with Lynch syndrome not displaying high penetrance in prostate cancer.2-4 The remainder of DDR defects seen in prostate cancer center mostly around the DNA double-strand break repair, replication stress signaling, and cell cycle regulation pathways. DDR gene alterations occur in ~25% of metastatic castration-resistant prostate cancer (mCRPC), with BRCA2 being by far the most frequently altered gene in this pathway, followed by ATM, and then to a lesser degree BRCA1 and CDK12, with more rare deleterious variants found in multiple other homologous recombination repair (HRR) and cell cycle genes.1 Alterations in BRCA2 are found significantly greater in advanced prostate cancer compared to primary disease, and certain histologic subtypes like ductal and cribriform disease are enriched for deleterious variants in DDR genes.5
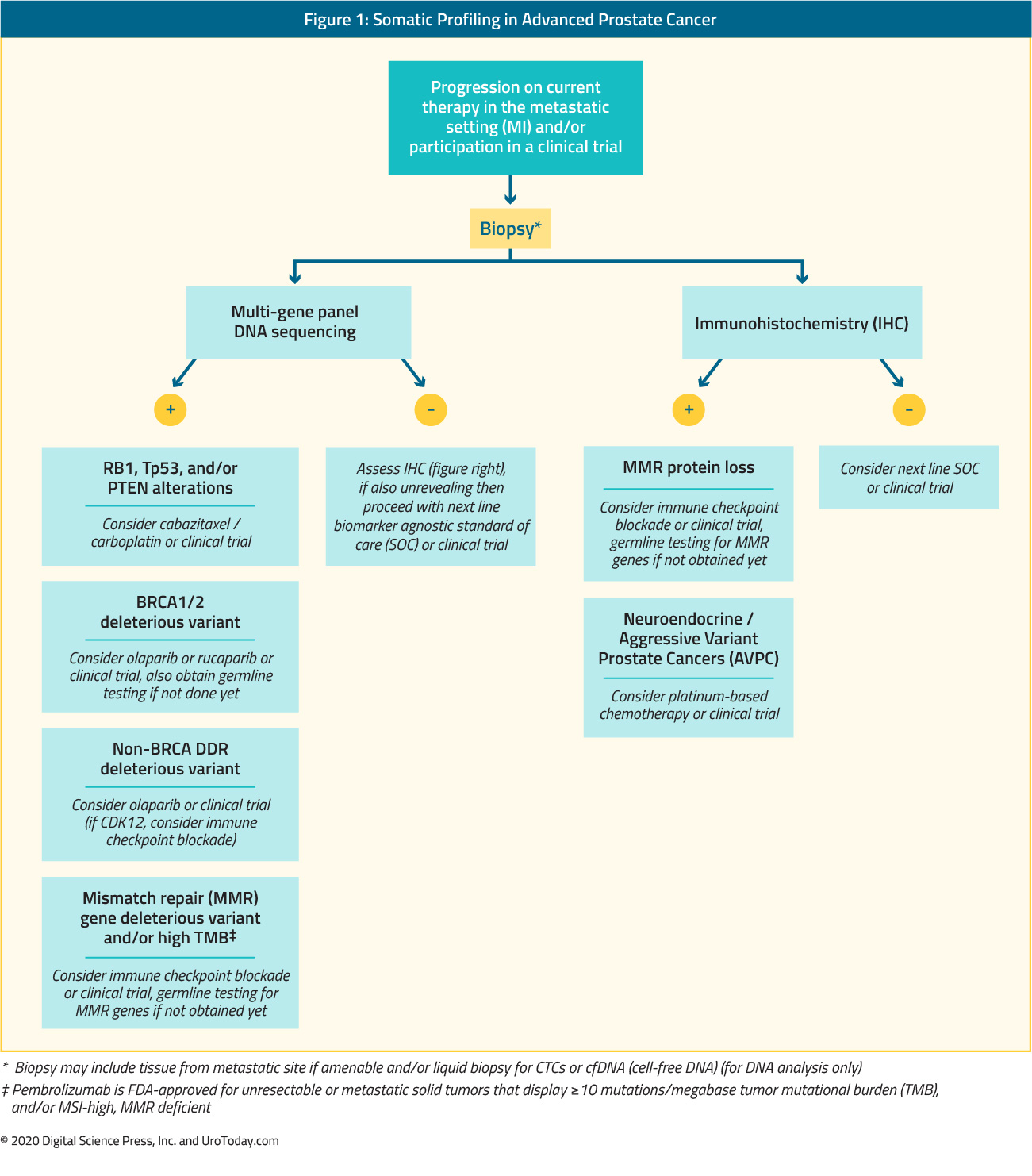
What are the modes of somatic testing for men with metastatic prostate cancer?
Somatic sequencing of prostate cancer cells from a patient can be performed on tumor tissue from a biopsy or surgical resection, and there are also a variety of “liquid” biopsy options, including circulating tumor cells and circulating tumor DNA. Unlike germline variants, the cancer’s genomic profile of DNA variants and associated altered pathways can evolve over time, with changes due to selective pressure from anti-cancer therapies, metabolic demands, microenvironment changes, and altered DNA repair and replication as that cancer grows and spreads.6 Thus the timing of when to request somatic sequencing on a patient’s prostate cancer should be based on when that sequencing will influence treatment decision making and/or participation in a clinical trial. Archived tumor material may not reflect the most up-to-date, potentially actionable variants for that patient’s cancer. Thus, ideally, treatment teams should consider obtaining a fresh biopsy of a progressing metastatic site (when technically feasible) or performing a liquid biopsy as close to the time of initiating therapy as possible. The sensitivity of liquid biopsies should be considered high enough to serve as a guide for therapy. Liquid biopsy approaches that are negative for DDR mutations can safely be considered negative.7
What are the possible results from targeted-panel clinical somatic sequencing?
Multiple approved tumor and liquid somatic sequencing platforms are available to clinicians. Each platform may slightly differ in terms of the genes included on the panel, as well as their coverage. If a variant is discovered in a gene included on that particular panel, the specific DNA sequence and subsequent amino acid change resulting from that variant are reported, along with the variant type (i.e. missense, frameshift, deletion, splice site). The functional and clinical consequences of that variant can be classified as a pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign, or benign, with published criteria, multiple online resources, and in silico tools for variant interpretation.8-10 The majority of variants identified from multi-gene panel somatic sequencing are missense VUS. Many DDR genes, such as ATM and others, are large genes with relatively few hotspot mutations, thus VUS continue to be a major issue for clinical sequencing across all cancer subtypes. There is a great need to incorporate clinical outcomes data with assays of biological function to better characterize VUS and ultimately better guide treatment selection for patients.
A genetic counselor is the most ideal member of the healthcare team to explain the meaning of a VUS in a cancer-related gene to a patient, and to update that patient and/or their family should the variant’s function be further delineated at a later date. Currently, there are multiple resources for treating clinicians to attempt to get further information on a variant’s function.11-13 However, it is important to note that a true VUS should not be used to guide treatment selection at this time.
Should a patient with somatic variant discovered by CLIA-approved next-generation sequencing (NGS) undergo clinical germline sequencing?
As discussed above, variants in DDR genes are seen in upwards of 25% of metastatic prostate cancer. As per national guidelines, all men with metastatic prostate cancer are recommended to undergo clinical germline sequencing of select DDR-related genes, with the strongest consensus for testing BRCA2, regardless of somatic sequencing results. In addition, given relatively high germline-to-somatic ratio and cancer-risk implications of deleterious variants in BRCA1/BRCA2 and Lynch syndrome genes (MLH1, MSH2, MSH6, PMS2), men who are discovered to have somatic variants in these genes, and who have not already undergone germline sequencing, should be referred for hereditary genetic testing with special attention to that specific somatic variant.14 However, variants in other common DDR genes, such as ATM and CHEK2, are often VUS, have moderate or low penetrance in prostate cancer, and display lower germline-to-somatic ratios, thus a somatic variant in one of these genes alone should not necessarily prompt hereditary testing. However, if the variant allele frequency is relatively high and/or if a patient displays a personal or family history concerning for a hereditary cancer syndrome, then germline testing may be considered.
What somatic variants currently have associated approved therapies for the treatment of men with prostate cancer?
Currently, there are three FDA-approved therapies for advanced prostate cancer with associated DNA-based biomarkers for patient selection and those include pembrolizumab, rucaparib, and olaparib. Briefly, pembrolizumab, a well-known immune checkpoint blockade therapy, was FDA-approved for treating advanced MMR deficient/MSI-high cancers, agnostic of tissue origin.15,16 Testing methods for identifying these cancers include MMR protein staining, MMR gene sequencing, MSI PCR testing, and tumor mutational burden via NGS. Rucaparib and olaparib, two drugs in the class of poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) were both recently approved by the FDA for biomarker-selected, previously treated advanced prostate cancer.17,18 Rucaparib was granted accelerated approval for men with mCRPC with deleterious variants in BRCA1 or BRCA2 only (somatic or germline origin) based on data from the ongoing TRITON2 study (NCT02952534) showing promising objective response rate (ORR) and duration of response (DOR) for rucaparib 600mg BID, with a Phase III confirmatory trial pending. Olaparib was approved for men with advanced prostate cancer who previously progressed on abiraterone or enzalutamide and have a deleterious HRR gene variant (somatic or germline) based on statistically significant improvement in radiographic progression-free survival (rPFS) compared to the alternative androgen-signaling directed agent, with statistically significant benefit in overall survival (OS) and ORR also seen in cohort A of the study which included men with BRCA1, BRCA2, and/or ATM variants.19 FoundationOne CDx™ (Foundation Medicine, Inc.) for selection of patients with mCRPC carrying HRR gene alterations and BRACAnalysis CDx® test (Myriad Genetic Laboratories, Inc.) for selection of patients with mCRPC carrying germline BRCA1/2 alterations were also approved as companion diagnostics. However, it is important to note that the identification of a deleterious germline or somatic variant in a DDR gene for patient selection for targeted therapy can be pursued with any CLIA/clinically-approved germline and/or somatic sequencing platform, as a gene mutation cannot be patented as previously decided in the 2013 supreme court case of Association for Molecular Pathology v. Myriad Genetics, Inc.
CDK12 has previously been implicated in regulating the expression of HRR genes, and loss of function in CDK12 has been shown to result in a distinct immunophenotype in prostate cancer.20,21 Thus, CDK12 alterations held promise as a potential predictive biomarker for PARP inhibition as well as immune checkpoint blockade. However, clinical data thus far has been mixed, where CDK12 alterations more clearly portend for worse outcomes to both AR directed therapy and chemotherapy but have yet to show clear benefit to PARPi or immune therapy.22-24
Are there differences in functional impact or outcomes based on if a patient has a BRCA2 variant of germline versus somatic origin?
Although mutations in BRCA1 or BRCA2 can be found across a variety of cancer subtypes, it is not clear that variants in these genes are ubiquitous “drivers” in each cancer subtype nor if they are targetable in a tissue-agnostic fashion. Recent data suggest that selective pressure for BRCA biallelic loss, true phenotypic impact, and subsequent therapeutic vulnerability to PARP inhibition are limited to those cancer subtypes where hereditary variants in BRCA1/2 confer an increased risk of that cancer (prostate, breast, ovary, and pancreas).25 Interestingly, in this study prostate cancer was the only BRCA-associated cancer with relatively equivalent germline to somatic BRCA-variants, and bi-allelic loss of BRCA in prostate cancer was seen to the same degree whether the variant was of somatic or germline origin.25 In the treatment of advanced prostate cancer, improved response to PARP inhibition has been noted regardless of BRCA1/2-variant origin, and emerging data demonstrate that responses can be seen even with just haploinsufficiency.19,25
There are multiple PARP inhibitors now FDA approved in various clinical settings or in late-stage clinical development. Are all PARP inhibitors equal in terms of biological action, side effects, and efficacy?
Preclinical and clinical data so far on the different PARPis have revealed many similarities, but also notable differences, most notably the chemical structure and size of each PARPi, that impact the drug’s potency, dosing, side effect profiles, and potentially patient biomarker selection. The PARP family of proteins plays a key role in a variety of cellular processes that includes DNA repair, chromatin modulation, transcription regulation, and aspects of the replication stress response. With regard to DNA repair, PARP1 and PARP2 play critical roles in DNA strand break repair through multiple DDR pathways, with HRR-deficient cells showing a greater reliance on PARP activity to maintain cell survival. PARP1 activity both opens up chromatin and facilitates the recruitment of downstream DNA repair factors to damaged sites. After completing its recruitment tasks, PARP is then released from its bound DNA via PARP auto-PARylation. Thus, the binding of PARP to damaged sites, its catalytic activity, and its eventual release from DNA are all necessary steps for a cancer cell to respond to DNA breaks introduced by certain chemotherapies, radiation, and various forms of endogenous damage. 26
All PARPis block PARP’s activity similarly; however, the preclinical potency and clinical dosing in patients are quite distinct between different PARP inhibitors. These differences are thought to center mostly on the ability of the inhibitor to block the release of PARP bound to chromatin, also termed its “PARP-trapping” capability. For example, talazoparib is able to bind chromatin and create these trapped PARP–DNA complexes to an approximately 100-fold greater degree than rucaparib, niraparib, or olaparib. Veliparib on the other end of the spectrum displays negligible PARP-trapping ability. In addition, preclinical models across various cancer types have shown that the cytotoxic potential of each of the clinically available PARP inhibitors differs between HRR-deficient (HRD) and HRR-proficient isogenic cell lines, whereby olaparib showed the most HRD-specific sensitivity, and talazoparib was the most agnostic of HRD status.26,27 While there has been no clinical data directly comparing the various PARP inhibitors to each other, there are differences in the incidence of adverse events (AE) seen in late-stage phase trials. For example, while all PARP inhibitors can suppress blood counts in patients, late-stage trials of PARPi for patients with various advanced solid cancers have shown grade >3 or higher anemia occurring in ~40% of patients with talazoparib versus ~20% with olaparib, while ~34% patients treated on niraparib in late-phase trials had grade > 3 thrombocytopenia.26 There has not been, nor is there likely to be, direct head to head trials of the various PARP inhibitors; however, preclinical data and differences in PARP-trapping ability may suggest the potential for differences in efficacy based on a patient’s biomarker status and tissue type.
Are there differences in outcomes in men with BRCA2 variants versus other DDR variants with respect to PARPi?
As noted above, rucaparib is currently approved only for men with mCRPC with deleterious BRCA1/2 variants, while olaparib received approval for a broader list of deleterious HRR genes. However, multiple subgroup analyses of clinical trials and retrospective analyses of men treated with PARPi have shown that responses, and particularly meaningful radiographic responses, are predominantly driven by those men with BRCA1/2 alterations.19,24,28,29 Meaningful responses have also been seen in the closely-related Partner and Localized of BRCA2 (PALB2) gene, though patients with variants in PALB2 are relatively rare in currently published studies. In converse, while there is a biological rationale for using PARPi in ATM-altered cancers and early phase in-human studies showed initial promise, relatively few meaningful responses to PARPi have been reported for men with ATM alterations from subsequent later stage studies and retrospective analyses, though some PSA response and reduction in CTCs have been seen in this patient population.24,30 However, loss of ATM may serve as a predictive biomarker for inhibitors of ATR, with multiple currently in clinical trials, with Phase I data showing promising safety and efficacy signals.31,32 A similar lack of significant response to PARPi has also been seen in an analysis of men with alterations in CDK12 as mentioned above.22 It is important to note that in any single prospective trial, the relatively small numbers of non-BRCA variant carriers combined with the relatively large smattering of genes included in the HRR pathway as associated biomarkers make finding significance in subgroup analyses of each individual gene not possible. Also, it is not known if responses may differ in patients with these alterations if PARPi is used earlier in a patient’s treatment course. A gene variant does not always reflect the active DDR signaling and function in a cancer cell, and prior treatments can influence these functions and reliance on alternative DDR pathways. Multiple more functional biomarkers are in development including genomic scars and transcriptomic signatures of DDR deficiencies, protein staining, and larger-scale proteomics of DDR genes, amongst others.31 In addition, biologically rationale combination treatment strategies, such as combined androgen-directed therapy with PARPi, may extend the benefits of PARPi to HRR gene wild-type patients, whereby androgen blockade can induce a BRCAness state and susceptibility to DDR inhibition. The recent Phase II trial of abiraterone plus olaparib versus abiraterone alone indeed showed rPFS benefit extended to the entire study population, regardless of HRR gene status.32 Further cooperative, correlative-rich studies in men with prostate cancer and subsequent data sharing will help facilitate the parsing out of each gene pathway’s impact on DDR function and integrated, adaptive biomarkers for selecting treatment with PARPi, platinum chemotherapy, other DDR inhibitors in development, and even potentially immune therapy for our patients.
Written by: Patrick G. Pilié, MD, Assistant Professor, Department of Genitourinary Medical Oncology, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, Texas
Published June, 2020
Related Content:
- PARP Inhibitors, Prostate Cancer and a Promise Fulfilled
- Germline Testing for DNA Repair Mutations in Prostate Cancer: Who, When and How?
- PARP Inhibitors - A Breakthrough in Targeted Therapies for Prostate Cancer
- DNA Repair and PARP Inhibitor Therapy in Prostate Cancer - Veda Giri, Patrick Pilié, and Arpit Rao