(UroToday.com) The Society of Nuclear Medicine & Molecular Imaging (SNMMI) 2021 Annual Meeting included a session on FDA Radiopharmaceutical updates and a presentation by Drs. Shane Masters and Sue Jane Wang discussing clinical trial design and statistical considerations for PSMA PET drug development. Dr. Masters notes that “Guidances by the FDA” are the FDA’s current thinking on a topic and are not legally binding documents. Furthermore, a ‘Guidance for Industry’ includes both academic and commercial sponsors.
Dr. Masters highlighted that indications for medical imaging agents are delineated by four categories, including (i) structure delineation, (ii) functional, physiological, or biochemical assessment, (iii) disease or pathology detection or assessment, and (iv) diagnostic or therapeutic management, for which the FDA “suggest that it would not be sufficient simply to demonstrate that the results of the test drug were used to direct a change in patient management, even to an intervention that is well established. Rather, we recommend that the sponsor establish whether the change was better or worse for the patient.”
To date, the PSMA PET drug is indicated for positron emission tomography (PET) of prostate-specific membrane antigen (PSMA) positive lesions in men with prostate cancer with (i) suspected metastasis who are candidates for initial definitive therapy, or (ii) suspected recurrence based on elevated serum PSA level. Dr. Masters notes that the regulatory standard of effectiveness is substantial evidence of effectiveness defined as “evidence consisting of adequate and well-controlled investigations, including clinical investigations”. This defines the type and amount of evidence, with the FDA trying to be flexible within regulatory limits. As follows is substantial evidence of effectiveness for PSMA-PET: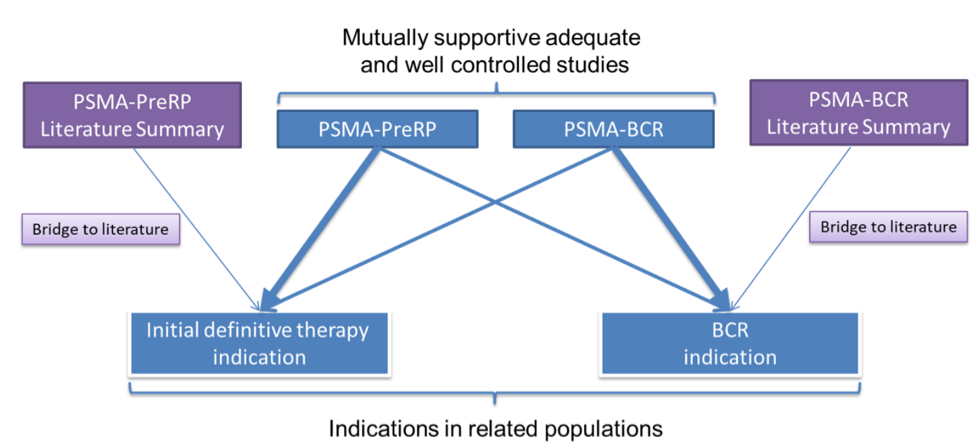
Dr. Masters then discussed several design considerations for phase 3 efficacy trials.
Selecting Subjects
For phase 3 trials, patients should be representative of the population in which the drug is intended to be used, with patient characteristics often reflected in the indication statement. For example, for PSMA PET/CT prior to radical prostatectomy, patients should have biopsy-proven adenocarcinoma, intermediate or high-risk disease, and planning to undergo a prostatectomy with pelvic lymph node dissection. Another example is for PSMA PET/CT in the biochemical recurrence setting. The patient should have histopathological proven prostate adenocarcinoma, a rising PSA after definitive therapy with prostatectomy or radiation therapy, with a threshold based on the AUA or ASTRO-Phoenix definition.
Imaging Conditions
The dose, dose to scan time, imaging protocol, criteria for image interpretation, etc should be optimized early in the development, prior to phase 3 efficacy trials, all of which should be included in the labeling. For example, for Ga-68 PSMA-11, image acquisition should begin at the proximal things and proceed cranially, and unless contraindicated, a diuretic expected to act within the uptake time period may be administered at the time of the radiotracer injection to potentially decrease artifact from radiotracer accumulation in the urinary bladder and ureters. Another example is for piflufolastat F 18 (PYLARIFY), the recommended start time for image acquisition is 60 minutes after PYLARIFY injection, and starting image acquisition more than 90 minutes after injection may adversely impact PYLARIFY performance.
Image Evaluations
The FDA recommends that image evaluators be fully blinded or blinded to the outcome (ie. not blinded to certain patient-specific information) and that any elements that are unblinded should be prospectively specified and defined in the protocol. Independence is also important and it is recommended that readers are unaware of the findings of other readers. Furthermore, it is much easier to maintain blinding when offsite image evaluation is performed, and at least 3 blinded, independent, central readers is the desired number.
Reference Standards
The FDA notes that “diagnostic standards may not be error-free”, but for purposes of a clinical trial, they are generally regarded as being definitive. Additionally, evaluation of the reference standard and investigational image evaluation should be independent, and evaluation with reference standards should be planned for all enrolled subjects. The decision to obtain reference standard data should be affected by investigational agent test results. For example, for initial definitive therapy, the reference standard is histopathology from pelvic lymph node dissection, and for biochemical reference it is a composite of histopathology when feasible, supplemented by post-PET imaging data and PSA level.
Endpoints
Success threshold endpoints should be predefined and the reader must meet the criteria for all primary endpoints to be successful. Furthermore, at least 2 of 3 readers should be successful for a positive study. For imaging trials, relying on sensitivity, specificity, positive predictive value, negative predictive value, and correct detection rate are commonplace: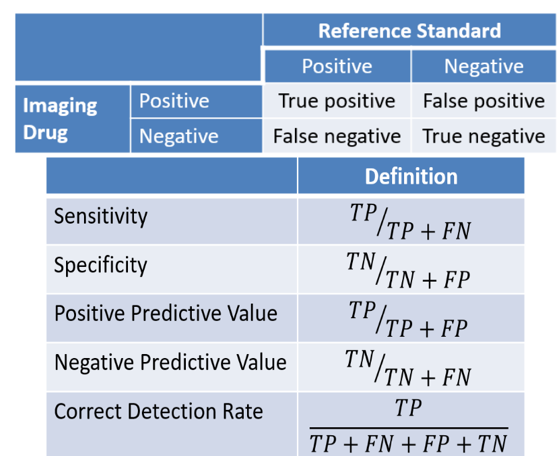
Dr. Wang then highlighted that when assessing diagnostic utilities of PSMA PET/CT, there are two target populations:
- Target population I (PSMA pre-radical prostatectomy): patients with intermediate to high-risk prostate cancer suspected of metastasis expected to undergo radical prostatectomy prior to initial definitive therapy (primary objective: diagnostic performance)
- Target population II (PSMA biochemical population): men with prostate cancer suspected of recurrence after prior therapy to localize metastatic disease based on elevated PSA levels (primary objective: diagnostic performance)
Important efficacy endpoints for target population I is patient-level sensitivity, specificity, and positive predictive value, whereas for target population II it is correct detection = number of true positives/number of scanned biochemical recurrence patients, as well as region-level positive predictive value, and patient-level positive predictive value. In general, there should be three readers blinded to histopathology and clinical follow-up data, with at least two readers achieving the success criteria (pre-specified threshold for each primary endpoint).
The following are two example questions of primary interest: 1) What is the diagnostic performance in the intent-to-image patients with primary prostate cancer receiving PSMA-PET planned to undergo radical prostatectomy prior to initial therapy? 2) What is the diagnostic performance in the intent-to-image patients with primary prostate cancer receiving PSMA-PET planned to undergo radical prostatectomy and whose histopathology is available. The concern with the second question is that histopathology may be selectively available/collected, such that this can easily lead to a biased assessment of the sensitivity and specificity of pre-radical prostatectomy patients. Furthermore, ICH E9 states that “the intention to treat principle implies that the primary analysis should include all randomized subjects. Compliance with is principle would necessitate complete follow-up of all randomized subjects for study outcomes.”
The Estimand Framework was an ICH E9(R1) Addendum “Estimands and Sensitivity Analysis in Clinical Trials published in 2019”. This is a structured framework to clarify treatment effects of interest, improving planning, design, analysis, and interpretation. Additionally, it facilitates dialogue between disciplines and between sponsor and regulator. The Estimand Framework is as follows: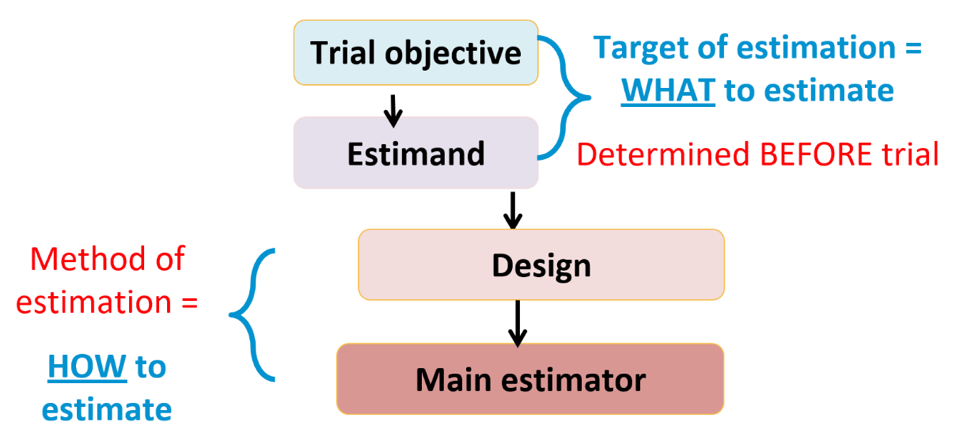
Dr. Masters and Wang concluded their presentation with the following take-home points:
- Estimands framework facilitates dialogue and helps ensure trials provide reliable answers to the right questions
- Implementation is important to public health in that E9(R1) provides a framework for discussion and advancement by dialogues between sponsor and review team seen in a few therapeutic developments
- Estimand framework for imaging drug developments is advancing trial efforts in progress
Presented by: Shane Masters, Division of Imaging and Radiation Medicine, Office of Specialty Medicine, Center for Drug Evaluation and Research, Food and Drug Administration, Silver Spring, MD
Sue Jane Wang, Deputy Director, Division of Biometrics I, Office of Biostatistics, Food and Drug Administration, Silver Spring, MD
Written by: Zachary Klaassen, MD, MSc – Urologic Oncologist, Assistant Professor of Urology, Georgia Cancer Center, Augusta University/Medical College of Georgia Twitter: @zklaassen_md at the Society of Nuclear Medicine & Molecular Imaging - 2021 Virtual Meeting, June 11-15, 2021