(UroToday.com) Tumor-only sequencing panels can capture many relevant pathogenic somatic mutations in patient tumors but also detect germline variants that are not represented in population-level germline variant databases and likely over-estimate tumor mutational burden (TMB)1. As many ethnic groups are under-represented in these population-level germline databases, the authors of this presentation hypothesized that TMB estimates in these patients would also be inflated.
To address this question, the authors utilized tumor-only exome sequencing from 701 patients with newly diagnosed multiple myeloma within the CoMMpass study and compared TMB calls with those from matched tumor-normal sequencing. 575 patients were identified as white patients, 126 were identified as black patients.
As shown in the figure below, tumor-identified variants were filtered in four ways for this study (1) by identified matched germline sequencing, or by minor allele frequency of variants in the 1000 genomes and ExAC databases at different thresholds (2) 0, (3) < 0.001, (4) < 0.01. While variant filtering using public databases inflated TMB calls at all thresholds when using information from all genes sequenced, the degree of inflation was higher in patients identified as black. Limiting TMB calls to OncoKB cancer genes only, a similar trend that was lesser in magnitude was observed at the looser minor allele frequency thresholds.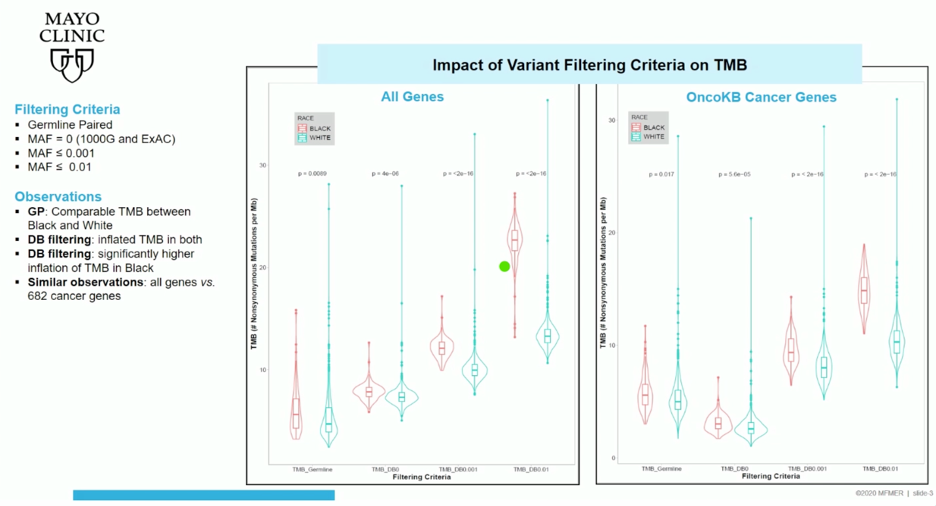
Similar results were obtained even when limiting the variants utilized from the 1000 genomes and ExAC databases to African and non-Finnish European ancestry.

These results highlight the importance of paired tumor-normal sequencing from the same patients to accurately quantify tumor mutational burden, especially for patients who are under-represented in available genomic databases, a critical issue in the context of recent approval of pembrolizumab immunotherapy based on a specific TMB threshold.
References:
1. Parikh K., Huether R.,, White K.,; et al., Tumor Mutational Burden From Tumor-Only Sequencing Compared With Germline Subtraction From Paired Tumor and Normal Specimens. JAMA Netw Open. 2020;3(2)
Presented by: Yan Asmann, Ph.D., Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN
Written by: Alok Tewari, MD, PhD, Medical Oncologist at the Dana-Farber Cancer Institute, at the 2020 European Society for Medical Oncology Virtual Congress (#ESMO20), September 19th-September 21st, 2020.