(UroToday.com) As prostate cancers harbor relatively low numbers of somatic mutations relative to other solid tumors, much research has been focused on identifying other molecular characteristics that explain disease phenotype. DNA methylation is an epigenetic mark, which means that it changes the effect of a DNA sequence without changing the sequence itself. This process is dictated in part by a balance of two classes of enzymes: (1) DNA methyltransferases, which place methyl groups on cytosine bases located next to guanine bases – also called CpG dinucleotides, and (2) demethylases such as TET enzymes – which hydroxylate methylcytosines – or cytidine deaminases such as APOBEC1 – which deaminate the methylcytosine and ultimately cause the replacement of the modified nucleotide with unmethylated cytosine.
In this presentation, Eric Small, MD, presented data on the landscape of DNA methylation in samples collected as part of the Prostate Cancer Foundation and Stand Up 2 Cancer (PCF/SU2C) West Coast Dream Team. This effort collected over 400 clinically annotated metastatic castration-resistant prostate cancer (mCRPC) samples for clinical and molecular analysis. To analyze DNA methylation, 100 mCRPC samples underwent whole-genome bisulfite sequencing, and data was integrated with RNA expression and DNA sequencing data.
This analysis revealed several findings:
- Disease progression from benign prostate tissue to mCRPC is associated with progressive hypomethylation of DNA globally
- Unsupervised analysis of methylation profiles revealed five clusters of samples
- One cluster identified treatment-associated small cell neuroendocrine prostate cancers, which has been previously described
- Another cluster showed hypermethylation (CMP phenotype), which was enriched for somatic mutations in genes such as TET2, IDH1, BRAF and DNMT3B. These mutations have been associated with CMP phenotypes in other malignancies such as AML and GBM. This CMP phenotype was identified in all biopsy sites, was not associated with particular clinical characteristics such as treatment response or PSA at diagnosis or with a difference in outcomes relative to other samples.
- Cluster 3 represented another relatively hypermethylated cluster.
- Cluster 3 and the CMP cluster had the highest overall survival relative to both the t-SCNC cluster, and the remaining other clusters (1 & 2)
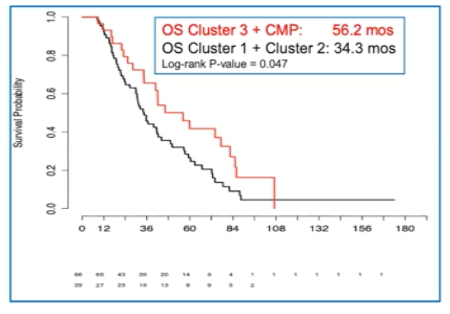
- A multivariate analysis for overall survival using PSA, LDH, hemoglobin, alkaline phosphatase, ECOG performance status, and presence of liver or visceral mets found that a methylation profile consistent with CMP or Cluster 3 was associated with a lower hazard ratio for death (HR 0.514, P=0.043)
In summary, this analysis identified two clusters of relatively hypermethylated patient samples (29% in total, CMP + Cluster 3) that were associated with improved overall survival relative to other samples. Therefore, Dr. Small proposed that mCRPC can be stratified by methylation pattern into 3 mutually exclusive prognostic groups: (1) Hypermethylated adenocarcinoma – best prognosis, (2) Non-hypermethylated adenocarcinoma, and (3) t-SCNC – worst prognosis. These findings are to be published in the near future.
Presented by: Eric Jay Small, MD, Professor of Medicine and Urology, Chief of the Division of Hematology/Oncology, University of California San Francisco, San Francisco, CA
Written by: Alok Tewari, MD, Ph.D., Medical Oncology Fellow at the Dana-Farber Cancer Institute, at the 2020 American Society of Clinical Oncology virtual annual meeting (#ASCO20), May 29th-May 31st, 2020 at the 2020 American Society of Clinical Oncology Virtual Annual Meeting (#ASCO20), May 29th-May 31st, 2020