Deleterious mutations in PBRM1 are frequent in cancer, especially in clear cell renal cell carcinoma where they are an early driver event occurring subsequent to VHL alteration in approximately 40% of patients – representing ≈ 175,000 new cases worldwide every year. Until recently, PBRM1 defects were often overlooked in clinical practice because there was no technology with sufficient sensitivity available to detect them. Despite recent advances made to better diagnose PBRM1 mutations, notably thanks to the advent of next-generation sequencing, patients with PBRM1-defective cancers (including but not restricted to clear cell renal cell carcinomas) still lack treatment options, which represents an important unmet medical need.
Polybromo 1, the protein encoded by PBRM1, is a subunit of one of the three forms of mammalian SWI/SNF complexes, the polybromo-associated BAF (PBAF) complex, which participates to the epigenetic process of chromatin remodeling. As its name indicates, PBRM1 comprises several bromodomains (BD1-6) that recognize acetylated lysine histone residues, mediating the chromatin targeting of PBAF complexes, especially at sites of active promoters that bear acetylated H3K27 and trimethylated H3K4 histone marks1 (Figure 1). PBRM1 acts as a tumor suppressor, notably by promoting genome stability through the maintenance of centromeric cohesion during mitosis. There is now growing evidence that PBRM1 is also involved in the DNA damage response: recent studies have indeed highlighted a role for PBRM1 in (i) promoting DNA replication re-priming downstream of stalled replication forks, and (ii) facilitating DNA repair at sites of DNA double-strand breaks through ATM-dependent silencing of transcription.2 Of note, PBRM1 also influences the anti-tumor immune response, but its role in shaping cancer cell immunogenicity remains controversial and poorly understood.3–6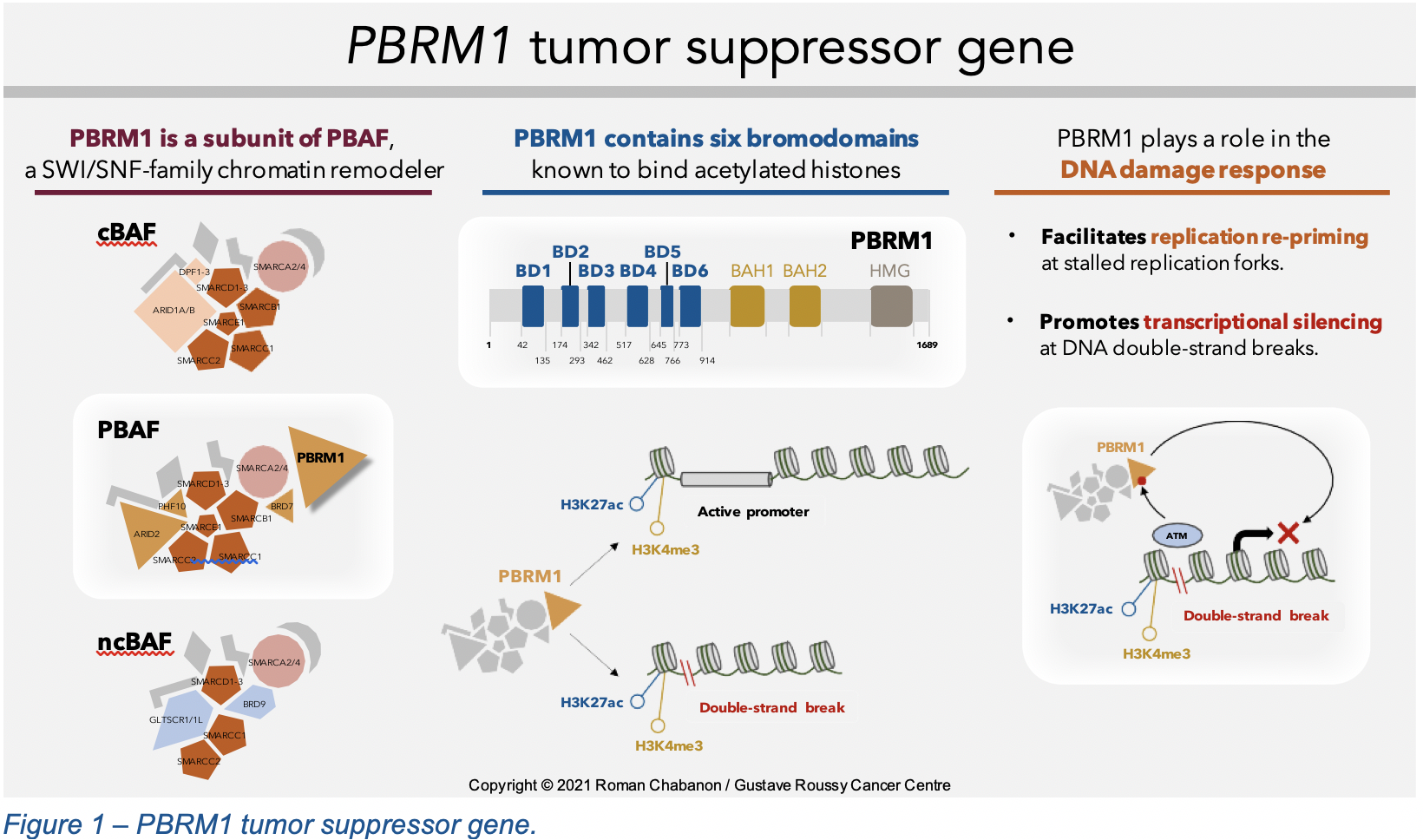
Because of the recently described roles of PBRM1 in the DNA damage response, we hypothesized that the loss of PBRM1 tumor suppressor gene function could be targeted using synthetic lethality, a concept that has already demonstrated its clinical efficacy in the context of BRCA gene-mutated breast and ovarian cancers, where PARP inhibitors are approved in the metastatic setting.7
To investigate novel therapeutic strategies for the treatment of PBRM1-defective cancers, we took a functional genomics approach based on the implementation of several complementary genetic perturbation and drug sensitivity screens (Figure 2). This identified previously undescribed synthetic lethal effects between the loss of PBRM1 and PARP inhibition, causing PBRM1-defective cancer cells to be approximately 10 to 100 times more sensitive to PARP inhibitors than cancer cells with an intact PBRM1 function8. A similar synthetic lethality was also identified with ATR inhibitors, another DNA repair-targeted therapy. We validated these observations in multiple isogenic and non-isogenic models from various histological subtypes, including clear cell renal cell carcinoma and non-small cell lung cancer. Considering the relative consistency of this phenotype across several models carrying different genetic backgrounds, this suggested that our findings might have translational utility in several cancer types where PBRM1 is mutated.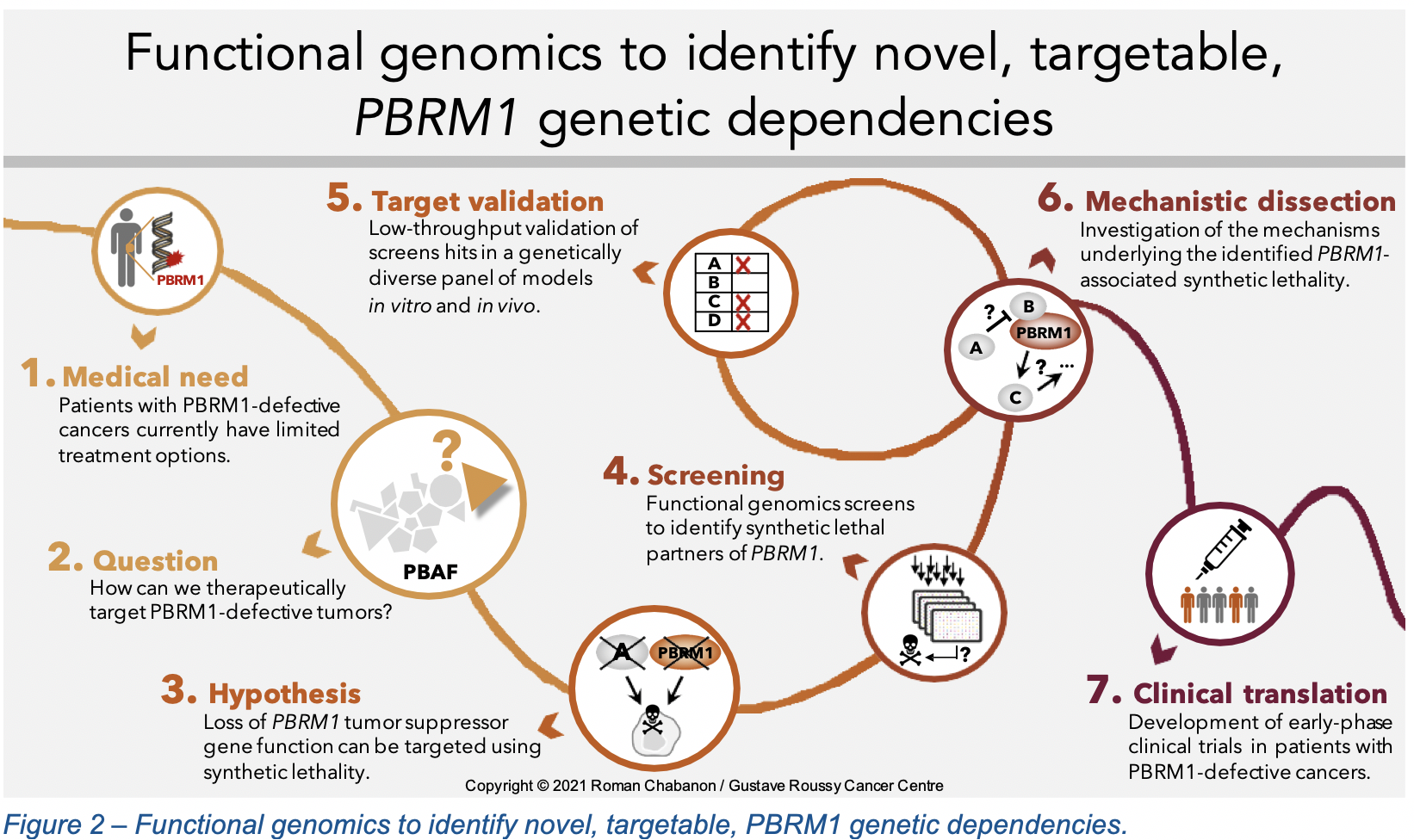
When further exploring the potential causes of this PBRM1/PARP inhibitor synthetic lethality, we found that the loss of PBRM1 associated with an elevated R-loop burden, increased replication stress, DNA damage and genomic instability. PBRM1-defective cancer cells were indeed characterized by an impaired progression of DNA replication forks, higher levels of γH2AX foci and the presence of micronuclei.8 In response to PARP inhibitor exposure, these genetic vulnerabilities resulted in an exacerbation of the pre-existing replication stress, and the accumulation of DNA damage and micronuclei to a degree that was no longer compatible with cell survival (Figure 3). Furthermore, we found that exposure of PBRM1-defective cells to PARP or ATR inhibitors led to a profound cell-autonomous activation of the innate immune signaling cGAS/STING pathway via the accumulation of cytosolic DNA in the form of micronuclei, which triggered a type I interferon response associated with the secretion of chemotactic and immunostimulatory cytokines.8 This finding is particularly important since immunotherapies have brought unprecedented benefits for patients with tumor types where PBRM1 is frequently mutated, such as clear cell renal cell carcinoma and non-small cell lung cancer. The activation of cGAS/STING signaling mediated by DNA repair inhibitors could therefore synergize with immune checkpoint blockade and further improve clinical benefit in patients with PBRM1-defective cancers.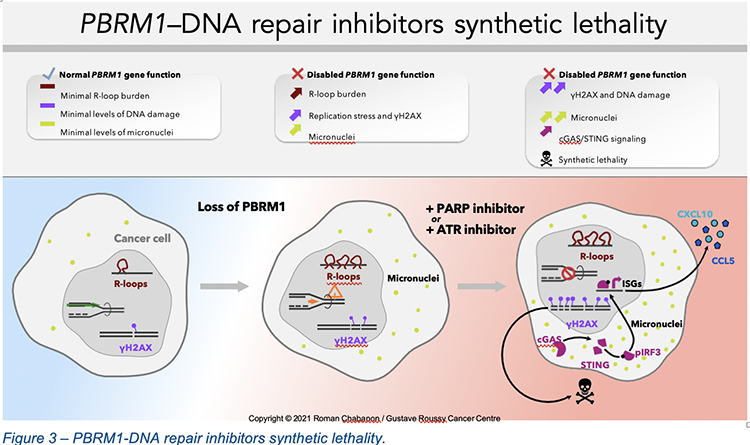
Theoretically, the three distinct forms of mSWI/SNF complexes assemble following an ordered, modular pathway,9 and have distinct functions and composition.1 PBRM1 is a specific subunit of the PBAF complex; it has no paralogs and is in fact the last subunit to be incorporated into PBAF. PBRM1 knockout has no effect on PBAF assembly, which supports our finding that the PBRM1/PARP inhibitor synthetic lethality is a direct consequence of PBRM1 loss. Importantly, this also underlines that the PBRM1/PARP inhibitor synthetic lethality is distinct from other genetic dependencies that have been recently described between DNA repair inhibitors and the cBAF-specific ARID1A subunit, or the ATPase SMARCA4 subunit.10–13 Therefore, our study describes for the first time that defects in a PBAF- specific subunit (namely PBRM1) lead to increased sensitivity to PARP and ATR inhibitors. This has important clinical implications, since PBRM1 defects occur in a clinically distinct patient population than ARID1A or SMARCA4 defects, where the use of PARP or ATR inhibitors has not previously been proposed.
Our findings now open novel perspectives for the treatment of patients with PBRM1-defective cancers, for whom no personalized medicine-based approach is available. Based on our findings, we are now exploring the PARP inhibitor plus immune checkpoint inhibitor combination therapy in molecularly-selected patients with clear cell renal cell carcinoma and other cancer histologies, in two ongoing investigator-initiated academic phase II clinical trials (ARIANES trial, NCT04276376; NIRADO trial, NCT04779151).
Written by: Roman M. Chabanon, PhD, Research Scientist. ATIP-Avenir group, Inserm Unit U981, Gustave Roussy, Villejuif, France. The CRUK Gene Function Laboratory, The Institute of Cancer Research, London, United Kingdom.
References:
- Michel, C. et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nature Cell Biology 20, 1410–1420 (2018).
- Chabanon, M., Morel, D. & Postel-Vinay, S. Exploiting epigenetic vulnerabilities in solid tumors: Novel therapeutic opportunities in the treatment of SWI/SNF-defective cancers. Seminars in Cancer Biology 61, 180–198 (2020).
- Miao, D. et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell Science 359, 801–806 (2018).
- Pan, D. et al. A major chromatin regulator determines resistance of tumor cells to T cell- mediated Science 359, 770–775 (2018).
- Bratslavsky, G. et al. PBRM1 mutation and immunotherapy efficacy: A comprehensive genomic profiling (CGP) Journal of Clinical Oncology 36, (2018).
- Zhou, et al. PBRM1 mutation and preliminary response to immune checkpoint blockade treatment in NSCLC. npj Precision Oncology 4, (2020).
- Lord, C. J. & Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158 (2017).
- Chabanon, R. M. et al. PBRM1 deficiency confers synthetic lethality to DNA repair inhibitors in Cancer Research [in press] (2021).
- Mashtalir, N. et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Cell 175, 1272–1288 (2018).
- Shen, J. et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Cancer discovery 5, 752–67 (2015).
- Williamson, T. et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nature Communications 7, 13837–13837 (2016).
- Kurashima, K. et al. SMARCA4 deficiency-associated heterochromatin induces intrinsic DNA replication stress and susceptibility to ATR inhibition in lung adenocarcinoma. NAR Cancer 2, zcaa005–zcaa005 (2020).
- Gupta, M. et al. BRG1 Loss Predisposes Lung Cancers to Replicative Stress and ATR Cancer Research 80, 3841–3854 (2020).