“Prostate cancer is one of the most commonly diagnosed cancers, affecting countless patients and families,” says lead author Jindan Yu, MD, PhD, an endowed chair professor of urology in the Emory School of Medicine. “While it often responds well to hormone therapy, many cases eventually develop resistance. One major pathway leading to treatment failure and disease progression is the transformation of prostate tumor cells into neuroendocrine prostate cancer (NEPC), a new beast that lacks the targets for existing prostate cancer drugs.”
The team mapped a stepwise process by which prostate cancer cells evolve into treatment-induced NEPC, an aggressive variant that behaves very differently from the original tumor. By showing that FOXA2 opens the door, NKX2-1 walks through it, and p300/CBP activates the new state, the authors give us both a model of how resistance emerges and a targetable point where we may be able to intervene before the disease becomes terminally neuroendocrine (Fig.1).

Figure 1. Cartoon illustrates the co-operation among FOXA2, NKX2-1, and p300 in driving chromatin remodeling of neuroendocrine prostate cancer. This figure was generated using Google’s Gemini model (Google AI, 2025). The content was reviewed and edited by the authors.
This is highly relevant clinically because treatment-induced NEPC is exactly what we see in a subset of men with advanced disease. They start with an androgen receptor (AR)–driven tumor, we treat them with potent AR-targeted agents, the AR pathway is suppressed, and then the cancer “escapes” by switching lineage. Once that happens, the tumor no longer depends on AR, starts using neural and developmental programs, grows faster, and becomes much less responsive to the therapies we rely on.
Mapping the Cells in 3D
Using high-depth Hi-C on patient-derived tumors and a cell line model, and in collaboration with Dr. Jonathan Zhao, associate professor in human genetics, the team developed the first-ever 3D map of how prostate cancer cells rewire themselves over time to become a new threat. This map illustrates how DNA folds into loops, compartments, and domains inside the cell nucleus. It also shows how enhancer-promoter connections are gained at neural regulators like the HOXB cluster and at important factors like NKX2-1, and are lost at luminal and AR-driven genes. The NEPC-specific loops are linked to genes involved in neuronal development and axonogenesis. These structural changes help switch on neuroendocrine gene programs and shut down prostate-lineage programs, which is exactly what drives the deadly transformation.
The researchers started from a simple observation: NEPC and castration-resistant adenocarcinoma (CRPC) have very similar genetic aberrations known as of today, so genetics alone cannot explain the switch. They profiled patient-derived xenografts of CRPC and NEPC with Hi-C and showed that the 3D genome architecture is different in the two states. NEPC tumors had thousands of chromatin loops that were not present in CRPC, and these NEPC-specific loops linked to genes involved in neuronal development and axonogenesis. CRPC, by contrast, kept loops around luminal/AR genes. So, the first message is: lineage plasticity in prostate cancer is an architectural event, not just a transcriptional one.
To model how this happens, they forced expression of FOXA2 into classic AR-positive LNCaP cells and watched the cells over 28 days. That alone was interesting: FOXA2 gradually pushed the cells from a luminal/AR state to a neuroendocrine-like state, both inducing and being propelled by dynamic chromatin architectural changes, ultimately resembling patient NEPC. This is important because it tells us the process unfolds in a stepwise and feedforward manner. Clinically, this fits what we see: patients do not flip to NEPC overnight.
The Role of FOXA2-NKX2-1 Axis
FOXA2 is the pioneer that opens neuroendocrine enhancers, but it alone is not enough to induce lineage change. FOXA2 first induces NKX2-1, a neural transcription factor that is normally not expressed in prostate epithelium. Once NKX2-1 is turned on, it binds mainly at promoters and, through chromatin looping, connects to FOXA2-bound enhancers. Together, they drive a steady NE transcription program by recruiting p300/CBP to catalyze H3K27 acetylation (Fig.2). Luminal markers remained, NE markers did not rise, and enhancer marks did not flip when the researchers knocked down NKX2-1, which prevented FOXA2 from completing the transition. However, the transition was significantly faster when they overexpressed both NKX2-1 and FOXA2. Of note, NKX2-1 was high in >50% of NE lesions, but some NEPCs were NKX2-1–high and FOXA2–low, meaning other pioneer factors (e.g., ASCL1) can feed into the same chromatin network.
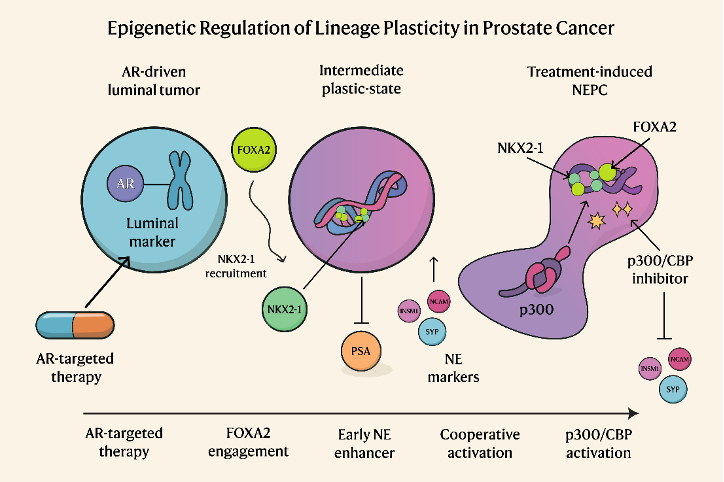
Figure 2. A diagram showcasing the stepwise process of neuroendocrine transformation driven by FOXA2 upregulation. This figure was designed by the authors using Inkscape v1.4.2 (Inkscape.org) illustration software.
How Would This Help Clinicians and Their Patients?
First, it gives us a more specific biomarker than “NE markers positive.” FOXA2 can be upregulated in several settings, but strong NKX2-1 in AR-low/NE-high tumors were much more specific in their tissue microarrays. That means NKX2-1 immunohistochemistry could help flag emerging NEPC in a male whose PSA is no longer tracking disease. In addition, epigenetic markers, such as H3K27ac and H3K4me1 at NE enhancers, might be useful to predict the lineage switch. Second, this model explains why some tumors with TP53/RB1 loss progress to NE, and some do not: if they cannot induce NKX2-1 (or an equivalent partner), the chromatin cannot complete the switch. That opens the door to patient stratification.
The p300/CBP component is the most translational point of this study. The researchers explored pharmacologic p300/CBP inhibition since NKX2-1/FOXA2 requires p300/CBP to activate NE enhancers. They found that this inhibition reduced NE gene expression and prevented NEPC growth both in vitro and in vivo. That is a clean, actionable vulnerability. We already worry that once AR is gone, we have nothing to target; this work says the epigenetic engine that keeps NEPC running is, in fact, druggable. It also suggests a timing idea: using p300/CBP inhibitors in patients who are showing molecular signs of lineage drift (AR↓, FOXA2↑, NKX2-1↑) might prevent full NE conversion.
In short, this study pulls together what has been scattered across different reports: AR pathway pressure → FOXA2 engagement → induction of NKX2-1 → cooperative looping with FOXA2 → p300/CBP–driven enhancer activation → stable NEPC. This research has major implications for patients, families, and the future of prostate cancer treatment. By identifying the molecular drivers of NEPC and showing how to block them, the study opens the door to new therapies that could dramatically improve outcomes.
Written by: Ramin Yousefpour Shahrivar, MS, Liu Peng, PhD, Jonathan Zhao, MD, and Jindan Yu, MD, PhD
- Emory University School of Medicine, Atlanta, GA