Heart disease and cancer are the leading causes of death in the United States.1 Prostate cancer (PC) is the most common cancer in American men, and PC is most frequently diagnosed among men aged 65 to 74 years.2 The American Cancer Society’s estimates for PC in the United States for 2017 are about 161,360 new cases. Of these, about 26,730 are expected to die of the disease.1
Cardiovascular disease (CVD) comprises all disorders of the heart and circulatory system, including coronary heart disease, peripheral arterial disease, cerebrovascular disease, cardiomyopathies, congenital heart disease, and other heart and circulation conditions. Nearly 801,000 deaths in the nation are accounted for by CVD annually (approximately 1 of every 3 deaths), with CVD being the leading cause of death for noncommunicable diseases.3 Coronary heart disease results in mortality in 45.1% of cases attributable to CVD in the United States, followed by stroke (16.5%), heart failure (8.5%), hypertension (9.1%), diseases of the arteries (3.2%), and other cardiovascular (CV) conditions.4,5
Pathophysiology
The most common underlying cause of CVD is atherosclerosis.6 When the process occurs in the arterial system, the vessels become narrow in a number of different vascular beds due to the gradual build-up of fat and cholesterol substances (atheroma or arterial plaques) within the walls of the arteries. Atherosclerosis can affect any artery in the body, including those in the heart, brain, arms, legs, pelvis, and kidneys. Consequently, various diseases may develop based on which arteries are affected. Multiple CV risk factors contribute to the development of arteriosclerosis, such as diabetes mellitus, smoking, hypertension, hyperlipidemia, obesity, inactivity, and inflammation. If atherosclerotic plaques are disrupted or eroded, thrombotic and embolic complications, including sudden cardiac death, acute myocardial infarction, acute stroke, and other manifestations, may ensue.
Epidemiology
The incidences of PC and of CV events increase with age. In a prospective cohort study, 22,048 men 40 to 84 years old, who were free of major disease, were followed from 1982 to 2007. In this study, the occurrences of new CVD and cancer climbed until age 80, and the CVD rate continued to rise after the 80th year. The incidence of carcinoma late in life apparently declined in cancers that are usually detected by screening, such as PC.7,8
Furthermore, the findings of two large cohort studies showed that CVD is the second leading cause of death after PC and other nonprostatic causes, regardless of the androgen deprivation therapy (ADT). One study was conducted by the South European Uroncological Group or SEUG of 766 patients to determine whether intermittent ADT is associated with a shorter time to progression,9 and the other study was a controlled one of 985 patients conducted by the European Organisation for Research and Treatment of Cancer or EORTC.10 ADT is being prescribed increasingly for the treatment of local or regional PC. An epidemiologic study of 37,443 population-based men who were diagnosed with local or regional PC in the Veterans Healthcare Administration concluded that ADT using a gonadotropin-releasing-hormone (GnRH)-agonist was associated with an increased risk of diabetes and CVD.11
In 2010, these findings led the U.S. Food and Drug Administration to require manufacturers of GnRH agonists to add extra safety information to drug labels to include the increased risk of diabetes and certain CV events (heart attack, sudden cardiac death, stroke) in men treated for this type of PC. The European Medicines Agency or EMA also prompted a similar label change for GnRH-agonists and GnRH-antagonists as follows, “Cardiovascular disease such as stroke and myocardial infarction has been reported in the medical literature in patients with androgen deprivation therapy. Therefore, all CV risk factors should be taken into account.”12
Methods of androgen deprivation therapy
ADT is pharmacologic hormonal manipulation. Normally, in the otherwise healthy male, when androgen levels in the body are low, GnRH stimulates the pituitary gland to produce luteinizing hormone (LH), which then stimulates the production of androgens (testosterone) by the testicles. In turn, androgens stimulate the development of PC cells. ADT reduces the level of androgens in circulation, thereby mitigating the growth of androgen-dependent PC cells.13
Orchiectomy is one surgical procedure used to remove the testicles. Subcapsular orchiectomy, a more acceptable surgical method, takes away only the tissue in the testicles that produce androgens. Both procedures are irreversible and are associated with morbidity and the risk of post-operative complications. Chemical castration is the use pharmacologic agents such as antiandrogens (eg, flutamide and bicalutamide) to block the binding of androgen to its receptor. The most commonly used pharmacologic agents are luteinizing hormone-releasing hormone (LHRH)-agonists which prevent the secretion of LH. LHRH-agonists, or LHRH analogues, are synthetic proteins structurally similar to LHRH and bind to the LHRH receptor in the pituitary gland. LHRH is synonymous with GnRH, therefor LHRH-agonists are also called GnRH-agonists. Herein, we will use the term GnRH. Androgen deprivation with a GnRH-agonist relies on the continued presence of high levels of the agonist to saturate the receptors in the pituitary gland, causing that gland to stop producing LH. Without LH stimulation, testosterone ceases to be produced from the testicles. Unlike orchiectomy, the effects of these drugs on androgen production are reversible. Once therapy is halted, testicular androgen production usually resumes. In contrast, GnRH-antagonists prevent GnRH from binding to its receptors in the pituitary gland, thereby halting the secretion of LH and causing the body’s androgen levels to drop. Unlike GnRH-agonists, GnRH-antagonists do not lead to a testosterone flare.13
The relative risk of CVD with GnRH/LHRH-agonists in observational studies and randomized, controlled trials (RCTs)
A cohort study of more than 30,000 PC patients revealed that pre-existing CVD increases the risk of death in men with PC who receive ADT, especially during the first six months of treatment. Moreover, differences in the type of preexisting CVD apparently had an effect. Earlier ischemic heart disease had only minimal impact on overall mortality, whereas patients with pre-existing stroke had higher mortality than those without ischemic heart disease or stroke.14
The putative mechanism underlying the association of ADT with elevated risks of CV events in patients with PC is speculative. But it is thought to be related to metabolic and immunomodulatory changes induced by ADT that destabilize previous atherosclerotic plaques among those with prior CVD and potentially accelerate the progression of atherosclerosis.
Men with PC who were on ADT were subjects in a cohort study of 41,362 men investigated for the risk of thromboembolic disease while accounting for known risk factors. This study analyzed 8 observational studies and 8 RCTs. When the results were compared from risk and timing of CVD after androgen deprivation by the three modalities of ADT, the major finding was that CVD-related events were highest within 6 months of ADT initiation in men with pre-existing CVD, regardless of the type of ADT. Men who experienced a CVD event within one year before the start of ADT were at the highest risk during the first 6 months of therapy when put up against those in the comparison cohort.15 Nevertheless, efforts to explain the relationship between androgen deprivation and CV events remain elusive, and there remains considerable disagreement on the relative risk of association between ADT and CVD with GnRH/LHRH-agonists in observational studies versus randomized clinical trials. All of the observational studies showed a relative risk greater than 1.0, whereas the results from the RCT mixed. 15
The relative risk of CV events differs among the many agents used in the treatment of PC for GnRH-agonists versus GnRH-antagonists, and several hypotheses exist to explain why an association between ADT and increased CV events occurs.16 One possibility is that potential cardioprotection provided by testosterone is interrupted by ADT. Another explanation is that circulating T cells with the capacity to express the GnRH receptor become activated and then infiltrate atherosclerotic plaques, causing fibrous cap disruption and plaque destabilization. GnRH-agonist–mediated immune activation may, therefore, be linked to CVD.
The potential role of the metabolic syndrome
Metabolic syndrome is a disorder of energy use and storage, named for the cluster of conditions that include increased blood pressure, high blood sugar, excess body fat around the waist, and abnormal cholesterol or triglyceride levels.17 The metabolic syndrome typically occurs in obesity and inactivity and is characterized by decreased levels of androgens and increased levels of low-density lipoprotein, triglycerides, and insulin. Testosterone is required for metabolic homeostasis, and its deficiency is associated with increased fat mass (in particular, central adiposity), reduced insulin sensitivity, and impaired glucose tolerance as well as elevated levels of triglycerides and cholesterol plus low high-density-lipoprotein-cholesterol. When testosterone activity is blocked by ADT as a treatment for PC, a metabolic syndrome-like state may develop, leading to a progression of underlying atherosclerosis.6,13,15
Hypothalamic-pituitary-gonadal (HPG) axis
Testicular testosterone secretion is regulated by the HPG axis (Figure 1),18 which is a convention to talk about individual endocrine glands as a single entity because they often act together. When metabolic processes are so closely interdependent, the individual pathways do not operate alone. A reaction in one pathway creates a compensatory effect in another. The HPG axis plays a critical part in the development and regulation of body homeostasis, most importantly in the reproductive and immune systems.18
GnRH is secreted from the hypothalamus by GnRH-expressing neurons. The anterior portion of the pituitary gland produces LH and follicle-stimulating hormone (FSH), and the gonads produce estrogen and testosterone. The hypothalamic GnRH stimulates the anterior pituitary gland to release LH and FSH, which stimulate testosterone release from the testes. Testosterone, in turn, stimulates PC cell proliferation through the action of its active metabolite, dihydrotestosterone, on androgen receptors in the prostate cells. Meanwhile, circulating testosterone exerts a negative feedback control on hypothalamic LH secretion.18
Mechanisms of action of GnRH-antagonists and GnRH-agonists differ. GnRH-agonists work by overstimulating GnRH receptors. These agonists stimulate GnRH receptors in cells of the anterior pituitary gland. Constant exposure to high-affinity stimulation leads to downregulation of pituitary receptors, inhibition of LH and FSH release, and a concurrent reduction in testosterone production. With time, overstimulation leads to desensitization. Consequently, production of LH, FSH, and testosterone is reduced. However, GnRH-agonists stimulate testosterone production before shutting it down. This initial testosterone surge can result in a transient increase (flare) in PC growth and in some patients, a worsening of symptoms, or flare. Only after an initial LH surge is testosterone suppression achieved, which delays reaching castration levels of testosterone and stimulates overproduction of testosterone for the first 30 days. Occasionally, patients may experience additional surges during long-term treatment on re-administration of GnRH-agonists, known as acute-on-chronic flare response or microsurge.18
On the other hand, GnRH-antagonists inhibit testosterone production directly. The antagonist is a competitive inhibitor of pituitary GnRH receptors. This blockade directly suppresses the secretion of LH and FSH and thereby reduces testosterone production by the testes without the initial stimulation of testosterone seen with GnRH-agonists..18
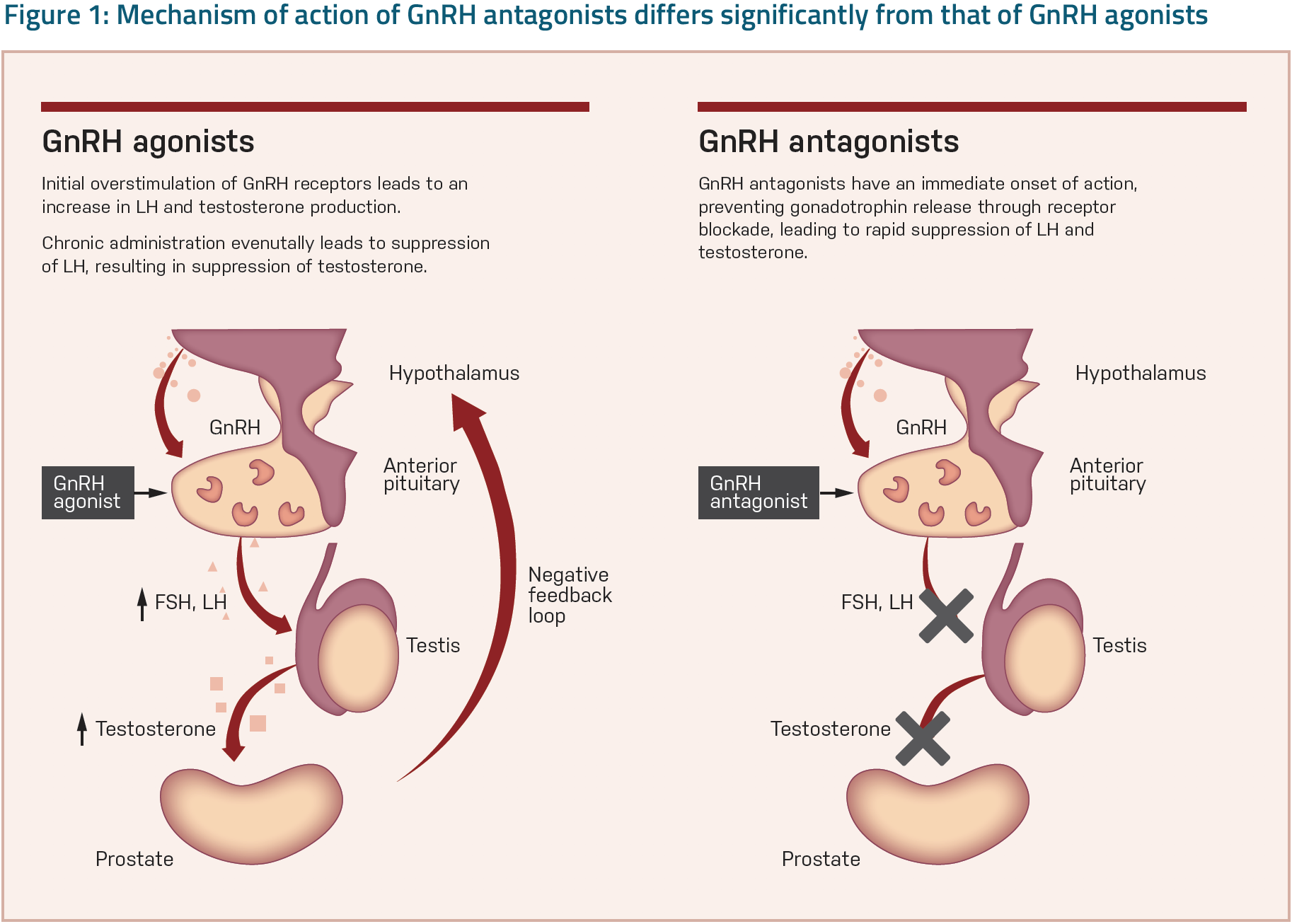
In RCTs, GnRH-antagonist therapy provides more rapid suppression of LH, FSH, and testosterone than GnRH-agonist treatment. Compared with the GnRH-agonist, there is evidence of improved disease control by a GnRH-antagonist, with a longer interval to prostate-specific-antigen progression and a greater reduction of serum alkaline phosphatase. In a post hoc analysis of six randomized trials, the risk of cardiac events within 1 year of initiating therapy was significantly lower among men receiving a GnRH-antagonist than a GnRH-agonist. Preclinical laboratory data suggest a number of mechanisms whereby GnRH-antagonist therapy may benefit men with pre-existing CVD. The most plausible hypothesis here is that unlike GnRH-agonists, GnRH-antagonists do not activate T-lymphocytes, which act to increase atherosclerotic plaque rupture.19
Conclusion
The putative deleterious effects of ADT on CVD progression and the risk of subsequent CV events underscore the need for individualizing patient treatment in light of key risk factors and a prior history of CVD. We have noted that the incidences of both CVD and PC escalate with older age, and CVD is the second leading cause of death among men with PC. Generally, ADT is associated with a higher risk of CV events, particularly in the first 6 months of treatment, in men with pre-existing CVD. Precisely why this occurs is speculative, but it may be related to metabolic and immunomodulatory changes that destabilize earlier atherosclerotic plaques and potentially accelerate the progression of underlying atherosclerosis. There is a potential hypothesis that GnRH-agonists stimulate T-cell–mediated proinflammatory responses, in turn leading to destabilization of atherosclerotic plaques compared with GnRH-antagonists. Such antagonists are associated with a reduced risk of CV events among men with pre-existing CVD compared with agonists. One credible hypothesis is that T-lymphocytes believed to contribute to atherosclerotic plaque rupture are not activated by GnRH-antagonists.
In PC, suppression of testosterone levels is central to the treatment goals for patients with that type of carcinoma. How this result is achieved needs to take into account the individual’s disease characteristics and any underlying conditions, especially CVD. The choice of ADT should thus be guided by the additional risk associated with those pre-existing conditions and the known characteristics of the ADT options.
Written by: Matthew T. Roe, MD, MHS, MHS, received his MD degree from Duke University School of Medicine from 1989-1993 and his MHS (Masters of Health Sciences in Clinical Research) degree in 2001 from the same institution. Dr. Roe completed an Internal Medicine residency at Duke University Medical Center from 1993-1996 and a Cardiovascular Fellowship at the Cleveland Clinic Foundation from 1996-1999. Dr. Roe was promoted to Associate Professor of Medicine, with Tenure, in the Division of Cardiovascular Medicine at Duke University Medical Center and the DCRI in January 2011.
References:
1. American Cancer Society. https://www.cancer.org/cancer/prostate-cancer/about/key-statistics.html
2. Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2013, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2013/, based on November 2015 SEER data submission, posted to the SEER web site, April 2016. SEER Stat Fact Sheets: Prostate Cancer. https://seer.cancer.gov/statfacts/html/prost.html
Accessed February 14, 2017.
3. National Heart, Lung, and Blood Institute. What Is Coronary Heart Disease? https://www.nhlbi.nih.gov/health/health-topics/topics/cad/
4. Heron M, Anderson RN. Changes in the Leading Cause of Death: Recent Patterns in Heart Disease and Cancer Mortality. US Dept of Health and Human Services Centers for Disease Control and Prevention, National Center for Health Statistics NCHS Data Brief. No. 254. August 2016. https://www.cdc.gov/nchs/products/databriefs/db254.htm
5. Benjamin EJ, Blaha MJ, Chiuve SE, et al; on behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2017 update: a report from the American Heart Association [published online ahead of print, January 25, 2017]. Circulation. doi: 10.1161/CIR.0000000000000485 /www.heart.org/idc/groups/ahamah public/@wcm/@sop/@smd/documents/downloadable/ucm_491265.pdf">https://www.heart.org/idc/groups/ahamah-public/@wcm/@sop/@smd/documents/downloadable/ucm_491265.pdf
6. National Heart, Lung, and Blood Institute. What is Atherosclerosis? https://www.nhlbi.nih.gov/health/health-topics/topics/atherosclerosis
7. Driver JA, Djoussé L, Logroscino G, et al. Incidence of cardiovascular disease and cancer in advanced age: prospective cohort study. BMJ. 2008;337:a2467.
8. Mozaffarian D, Benjamin EJ, Go AS, et al; on behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:e29-e322. //www.heart.org/idc/groups/heart-public/@wcm/@sop/@smd/documents/downloadable/ucm_472923.pdf">https://www.heart.org/idc/groups/heart-public/@wcm/@sop/@smd/documents/downloadable/ucm_472923.pdf
9. Calais da Silva FE, Bono AV, Whelan P, et al. Intermittent androgen deprivation for locally advanced and metastatic prostate cancer: results from a randomised phase 3 study of the South European Uroncological Group. Eur Urol. 2009;55(6):1269-1277.
10. Studer UE, Whelan P, Wimpissinger F, et al; EORTC Genitourinary Cancer Group. Differences in time to disease progression do not predict for cancer-specific survival in patients receiving immediate or deferred androgen-deprivation therapy for prostate cancer: final results of EORTC randomized trial 30891 with 12 years of follow-up. Eur Urol. 2014;66(5):829-838.
11. Keating NL, O'Malley AJ, Freedland SJ, Smith MR. Diabetes and cardiovascular disease during androgen deprivation therapy: observational study of veterans with prostate cancer. J Natl Cancer Inst. 2010;102(1):39-46.
12. Levine GN, D’Amico AV, Berger P, et al; American Heart Association Council on Clinical Cardiology and Council on Epidemiology and Prevention, the American Cancer Society, and the American Urological Association. Androgen-deprivation therapy in prostate cancer and cardiovascular risk: a science advisory from the American Heart Association, American Cancer Society, and American Urological Association: endorsed by the American Society for Radiation Oncology. Circulation. 2010;121(6):833-840.
13. National Cancer Institute Fact Sheet - Hormone Therapy for Prostate Cancer. https://www.cancer.gov/types/prostate/prostate-hormone-therapy-fact-sheet
14. Jespersen CG, Nørgaard M, Bjerklund Johansen TE, et al. The influence of cardiovascular morbidity on the prognosis in prostate cancer. Experience from a 12-year nationwide Danish population-based cohort study. BMC Cancer. 2011;11:519.
15. O’Farrell S, Sandström K, Garmo H, et al. Risk of thromboembolic disease in men with prostate cancer undergoing androgen deprivation therapy. BJU Int. 2016;118(3):391-398.
16. Veccia A, Maines F, Kinspergher S, et al. Cardiovascular toxicities of systemic treatments of prostate cancer. Nat Rev Urol. 2017 Jan 24. doi: 10.1038/nrurol.2016.273. [Epub ahead of print]
17. National Heart, Lung, and Blood Institute. What Is Metabolic Syndrome? https://www.nhlbi.nih.gov/health/health-topics/topics/ms
18. Mason M. Degarelix: a new hormonal treatment for prostate cancer. Future Prescriber. Vol 10, Issue 1, Version of Record online: 26 Feb 2009 Abstract.
19. Rosario DJ, Davey P, Green J, et al. The role of gonadotrophin-releasing hormone antagonists in the treatment of patients with advanced hormone-dependent prostate cancer in the UK. World J Urol. 2016;34(12):1601-1609.