(UroToday.com) Radiopharmaceutical Therapy Symposium (MRPTS) held in Palm Desert, California, was host to the session Unique Challenges with Clinical Trials for Radiopharmaceutical Therapy. Dr. Michael Morris discussed Industry Perspective and Regulatory Challenges for RPT Clinical Trials.
Dr. Morris began by outlining the biological rationale for targeting prostate-specific membrane antigen (PSMA), a highly expressed and therapeutically actionable surface protein in advanced prostate cancer. He emphasized that radioligand therapy (RLT) represents a convergence of molecular imaging and precision oncology, allowing for tumor-specific delivery of beta-emitting isotopes with meaningful antitumor activity in metastatic castration-resistant prostate cancer (mCRPC). A summary of the radiopharmaceutical pipeline was presented by Dr. Morris and is illustrated below.
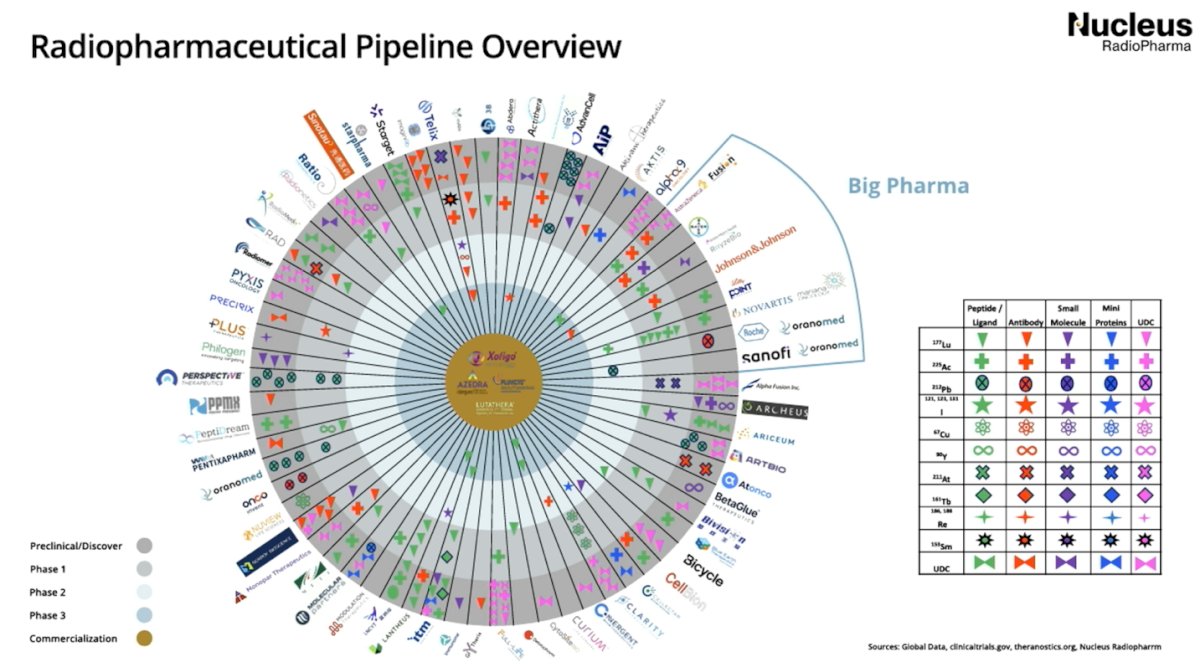
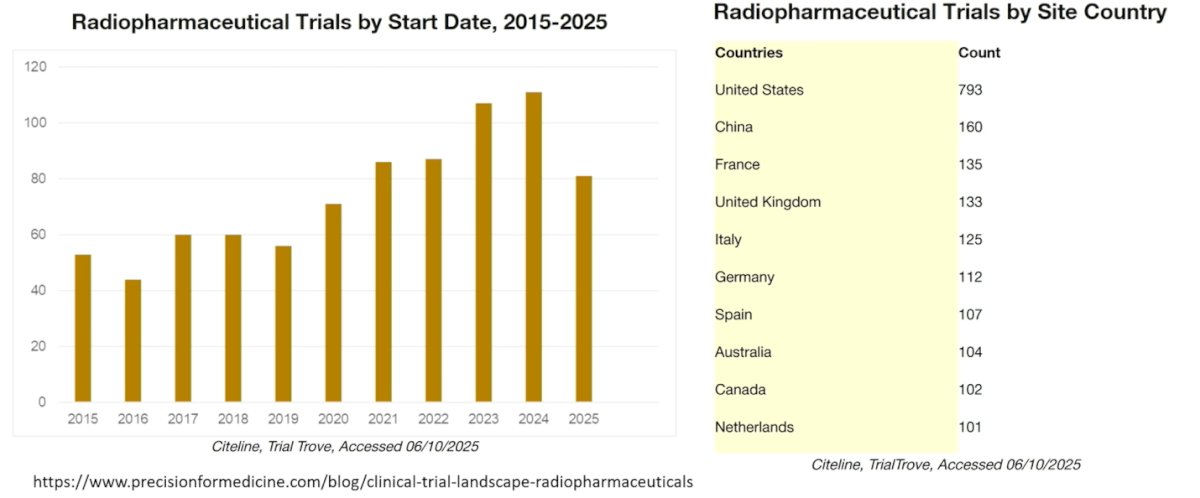
Dr. Morris showed that radiopharmaceutical clinical trials have increased steadily over the past decade, with marked growth beginning around 2020 and peaking in 2023–2024. The data highlight the rapid expansion of this therapeutic space, reflecting growing investment and clinical interest. Geographically, the United States leads by a wide margin in the number of active trial sites, followed by China and several European countries, including France, the United Kingdom, Italy, and Germany, underscoring the global momentum behind radiopharmaceutical development.
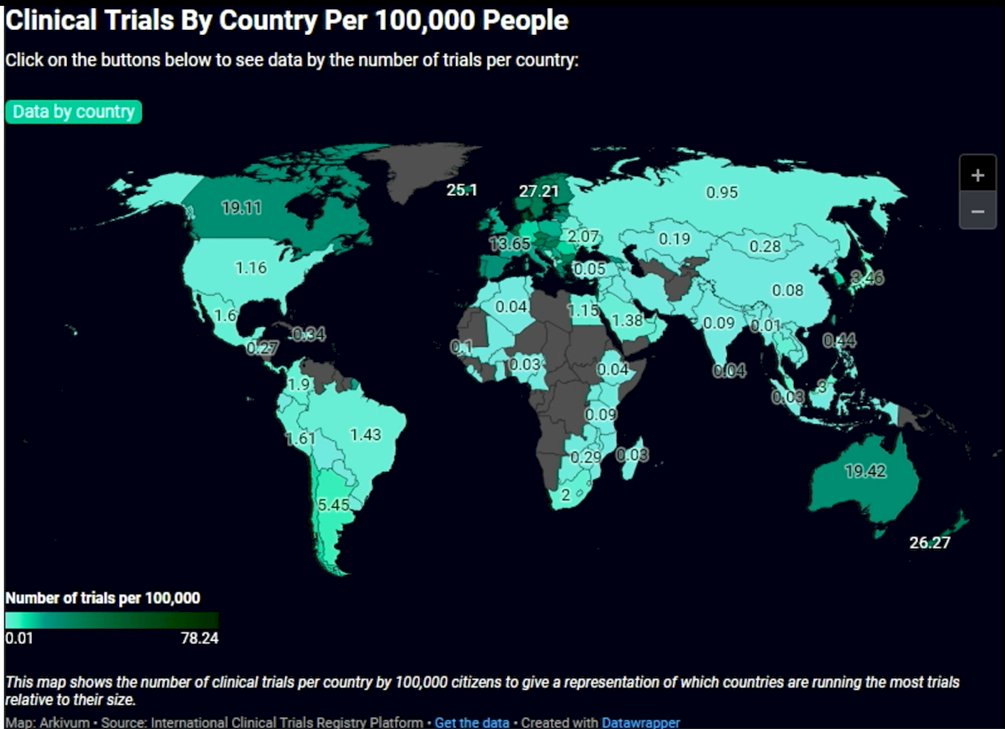
Moreover, when adjusted for population size, trial activity varies substantially by country. Several smaller or high-income nations demonstrate a disproportionately high number of trials per 100,000 people, including countries in Northern and Western Europe as well as Australia and New Zealand. In contrast, large-population countries such as the United States and China, while leading in absolute trial numbers, rank lower when normalized per capita. These data highlight important geographic disparities in research intensity and access to clinical trials worldwide.
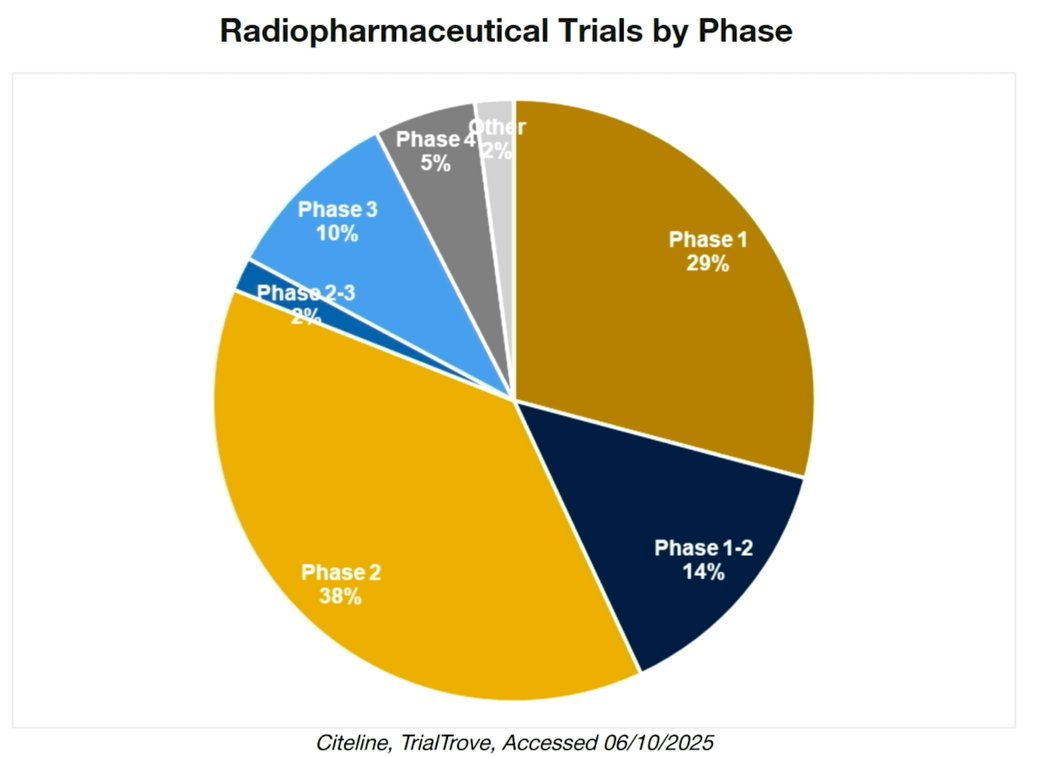
When examining radiopharmaceutical trials by phase, the majority remain in early development. Phase 2 studies account for the largest proportion (38%), followed by Phase 1 trials (29%) and Phase 1–2 studies (14%). Only 10% have reached Phase 3, with very small proportions in Phase 2–3 (2%) and Phase 4 (5%). These data underscore that most radiopharmaceutical development is still in the early- to mid-stage pipeline, with comparatively fewer agents advancing to late-phase, practice-changing trials.
Dr. Morris outlined the current FDA regulatory structure relevant to radiopharmaceutical development. Within the FDA’s Center for Drug Evaluation and Research (CDER), the Office of New Drugs oversees review activities, with radiopharmaceuticals intersecting two key groups. The Division of Imaging and Radiation Medicine (DIRM), under the Office of Specialty Medicine, is responsible for imaging and radiation-emitting medical products, with a focus on radiation physics and safety. In parallel, the Office of Oncologic Diseases, organized into disease-specific teams, regulates oncology drugs and biologics with an emphasis on clinical benefit and risk–benefit assessment. He also highlighted the Oncology Center of Excellence, created in 2017 to integrate oncology expertise across FDA centers, reflecting the increasingly multidisciplinary nature of cancer drug development.
Dr. Morris referenced recent discussions from the SNMMI Dose Optimization Workshop to emphasize that patient safety in phase 1 radiopharmaceutical trials must be framed within the broader clinical context. While radiation safety remains critical, the overall health risk varies substantially by disease stage. In metastatic prostate cancer, particularly in the castration-resistant setting where median survival may be measured in years or even months, undertreating the cancer often represents the greatest risk to patients. In contrast, for earlier-stage disease, where life expectancy can exceed a decade, long-term toxicities such as renal, hematologic, salivary gland injury, or secondary malignancies become increasingly relevant. These differences underscore the need to individualize dose optimization strategies according to disease stage and anticipated survival.
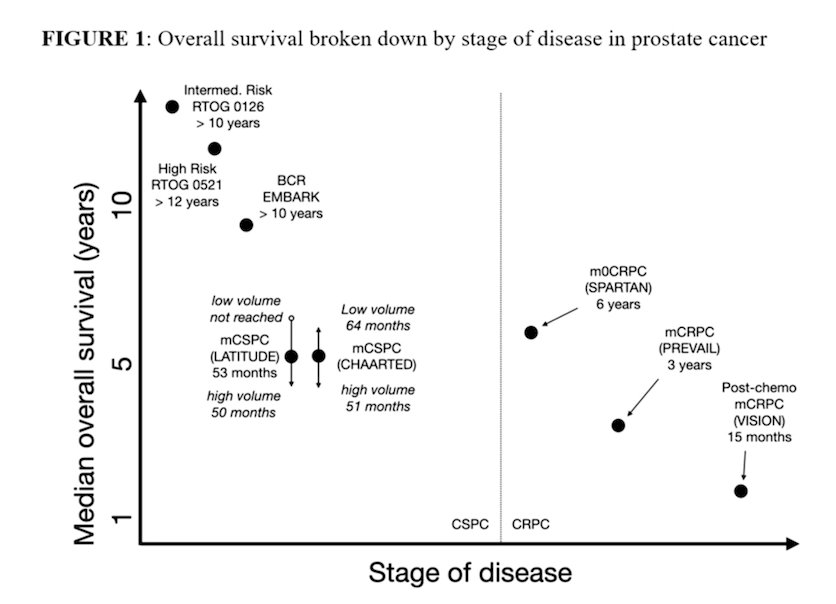
Moreover, he emphasized that dosimetry and safety considerations in radiopharmaceutical therapy should not simply mirror historical external beam radiation therapy (EBRT) constraints. Applying conventional EBRT organ dose limits to patients with advanced disease may be overly restrictive and not clinically appropriate. In fact, EBRT-based absorbed dose thresholds likely underestimate tolerable limits in the RPT setting and should not be used to arbitrarily cap dose escalation. Importantly, robust data directly linking absorbed dose to meaningful clinical toxicities remain limited. For this reason, well-designed phase 1 trials should be allowed to generate prospective safety and dose–response data to better define clinically relevant organ constraints in RPT development.
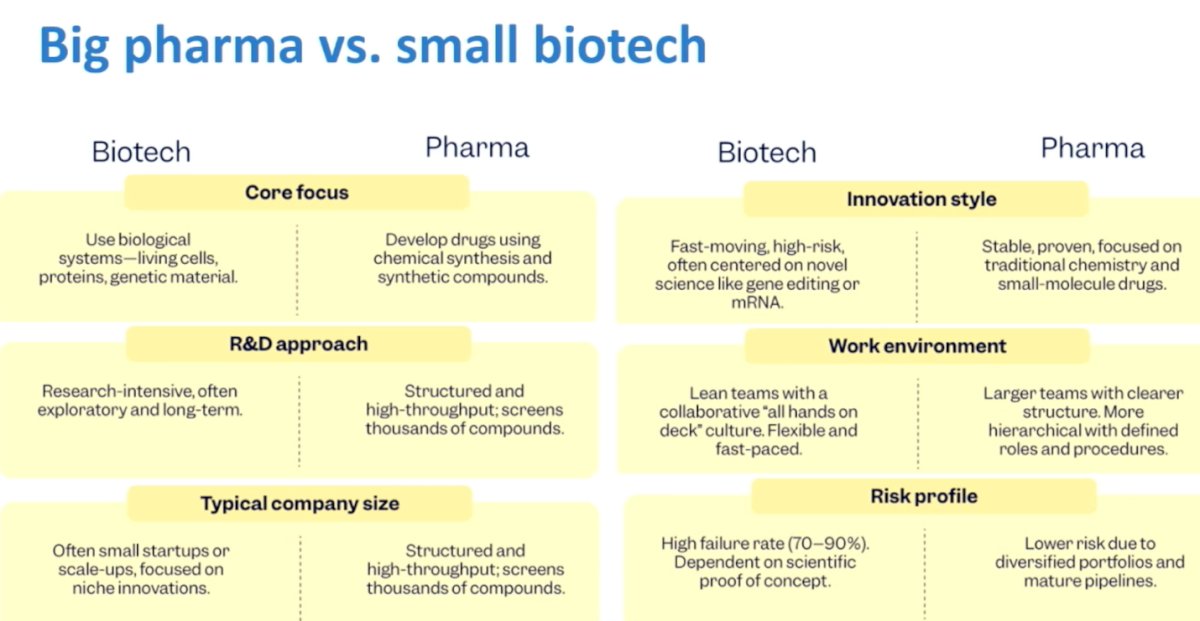
Dr. Morris emphasized the evolving dynamic between large pharmaceutical companies and smaller biotechnology firms in radiopharmaceutical development. Biotech companies often drive early innovation, operating with lean teams, higher risk tolerance, and a focus on novel platforms and biologically driven approaches. In contrast, large pharmaceutical companies tend to bring structure, scale, and resources, with established development pathways, broader portfolios, and the ability to execute large registration trials. While biotech frequently fuels scientific creativity and first-in-class concepts, pharma plays a critical role in late-stage development, global trial execution, regulatory navigation, and commercialization. Together, this ecosystem has accelerated progress in radiopharmaceutical therapy, but also introduces strategic, financial, and operational considerations that shape how programs move from early discovery to clinical adoption.
Furthermore, He went on to review the landmark VISION trial, which established the clinical benefit of 177Lu-PSMA-617 in patients with PSMA-positive mCRPC previously treated with androgen receptor pathway inhibitors and taxane chemotherapy. The addition of 177Lu-PSMA-617 to standard of care significantly improved overall survival and radiographic progression-free survival, firmly positioning PSMA-directed RLT as a new standard in the post-taxane setting. Subsequent trials, including PSMAfore and PSMA Addition, are exploring earlier lines of therapy, reflecting a broader strategic shift to integrate radioligand therapy across the prostate cancer continuum. (1)
Dr. Morris then transitioned to the broader technological and organizational journey behind Pluvicto’s development. He highlighted the early role of Endocyte, which secured rights to a promising PSMA-targeted compound and rapidly advanced development through focused collaboration, disease-specific expertise, and nimble decision-making. Endocyte’s structure allowed for rapid integration of scientific, regulatory, and clinical strategies, key elements in early-phase innovation.
Following acquisition by Novartis, the program entered a new phase characterized by global scale. With substantial financial, regulatory, and statistical infrastructure, Novartis executed multiple large international phase III trials and expanded the radioligand therapy platform. However, Dr. Morris noted that increased scale also brought added complexity. Decision-making became less direct, and the organization had to overcome initial challenges related to manufacturing, supply chain logistics, and global distribution—particularly critical for radiopharmaceuticals, where production timing and isotope half-life demand precise coordination.
Importantly, Novartis was relatively new to both prostate cancer therapeutics and radiopharmaceutical therapy at the time of acquisition, requiring rapid institutional learning and infrastructure development. Early manufacturing constraints underscored that clinical success alone is insufficient without parallel investment in production capacity and delivery systems.
Dr. Morris emphasized that the success of PSMA-targeted radioligand therapy reflects more than a single positive trial. It represents a multidisciplinary achievement spanning biology, imaging, nuclear medicine, medical oncology, regulatory science, and industrial logistics. He underscored that continued expansion of RLT will depend on optimizing patient selection, refining combination strategies, improving access to PSMA imaging, and ensuring equitable distribution of radiopharmaceutical infrastructure.
Dr. Morris concluded his presentation with the following key messages:
- RPT is rapidly expanding; while consolidation is likely, the field is currently experiencing an exceptional period of creativity, discovery, and innovation
- The U.S. regulatory environment for radioligand therapy is complex and evolving, generally posing challenges to early innovation but supporting the generation of high-quality data for approval
- Germany and Australia currently hold advantages in early discovery and exploratory development
- The United States maintains a strong advantage in conducting large, regulatory-grade trials
- The regulatory landscape continues to evolve across regions
- Industry is not monolithic; different development models serve distinct purposes across the drug development continuum
Presented by: Michael Morris, MD, Prostate Cancer Section Head, GU Oncology, Steven A. Greenberg Chair in Prostate Cancer Research, Memorial Sloan Kettering Cancer Center, New York, NY
Written by: Julian Chavarriaga, MD – Urologic Oncologist, Department of Urology at Penn State Health. @chavarriagaj on Twitter during the 2026 American Society for Radiation Oncology (ASTRO) Multidisciplinary Radiopharmaceutical Therapy Symposium, Palm Desert, CA, Tues, Feb 17 – Wed, Feb 18, 2026.
References: