(UroToday.com) The 2026 American Society for Radiation Oncology (ASTRO) Multidisciplinary Radiopharmaceutical Therapy Symposium (MRPTS) held in Palm Desert, California, between February 17th and 18th, 2026, was host to the session Unique Challenges with Clinical Trials for Radiopharmaceutical Therapy. Dr. Scott Tagawa discussed Radiopharmaceutical Therapy Clinical Trials: from an Investigator and Drug Development Perspective.
Dr. Tagawa began by outlining the learning objectives of the session, which focused on understanding the responsibilities of multidisciplinary investigators and their impact on clinical trial recruitment, recognizing the additional logistical requirements of physician-sponsored IND studies, including manufacturing, distribution, and funding oversight, and learning from real-world examples of prior and ongoing investigator-initiated trials. The audience was composed primarily of radiation oncologists (approximately 80%), with the remaining 20% representing pharmaceutical company representatives, reflecting the collaborative nature of radiopharmaceutical research and trial development.
Notably, 76% of attendees agreed that investigator-initiated or investigator-sponsored studies utilizing RPT are not easier to conduct than trials involving other therapeutic modalities. In addition, 54% of the audience had participated in RPT either as an investigator or sponsor, while 61% reported no prior experience obtaining an IND for an RPT study, underscoring the perceived regulatory and logistical complexity in this space.
Dr. Tagawa began by outlining his perspective as an academic GU medical oncologist actively involved in both therapeutic and biomarker-driven trials across multiple mechanisms of action, including investigator-initiated studies conducted with and without corporate or federal support, as well as industry-sponsored trials spanning phase 0 through phase 4. He emphasized that this broad experience provides insight into the unique opportunities and challenges associated with investigator-initiated trials (IITs) in radiopharmaceutical therapy (RPT).
He noted that IITs in RPT may offer greater opportunities for independent development compared to other therapeutic platforms. In some settings, obtaining an IND for RPT can be more straightforward, particularly when institutional expertise, infrastructure, and access to necessary components are already in place. The availability of similar base compounds for imaging and theranostic pairing further facilitates early-phase development, including phase 0 studies. However, he cautioned that despite these perceived advantages, successful execution still requires substantial knowledge, operational capability, and dedicated funding.
The development of β-radiolabeled J591 spans more than two decades, beginning with first-in-human studies demonstrating safety and tumor targeting in mCRPC. Early phase I and II trials established safety, dose-response relationships, and preliminary efficacy signals, including single-dose and fractionated dosing strategies. Subsequent expansion cohorts and combination studies with docetaxel confirmed feasibility and antitumor activity. More recently, randomized phase II data presented at ASCO GU 2023/2025 further support its clinical utility in combination with secondary hormonal therapy in low-volume, high-risk non-metastatic and metastatic castration-resistant prostate cancer, highlighting continued momentum in its clinical development.

Moreover, Dr. Tagawa discussed prior multicenter clinical trial experience, noting that when serving as sponsor, investigators may assume responsibility for manufacturing and distribution of the investigational product. A phase II trial of Lu-177–J591 was conducted between Weill Cornell Medicine and MSKCC, followed by a multicenter phase II study comparing Lu-177–J591 versus Indium-111–J591 across U.S. sites, with discussions of potential European expansion. These efforts highlighted key operational challenges, particularly related to radiopharmaceutical manufacturing, product stability, and transport logistics across participating centers.
Dr. Tagawa then reviewed the preclinical development of Actinium-225–J591. Actinium-225 nitrate was sourced from the Department of Energy and conjugated to DOTA-J591, followed by separation, sterilization, and quality control testing. Immunoreactivity and stability studies supported advancement toward an IND.
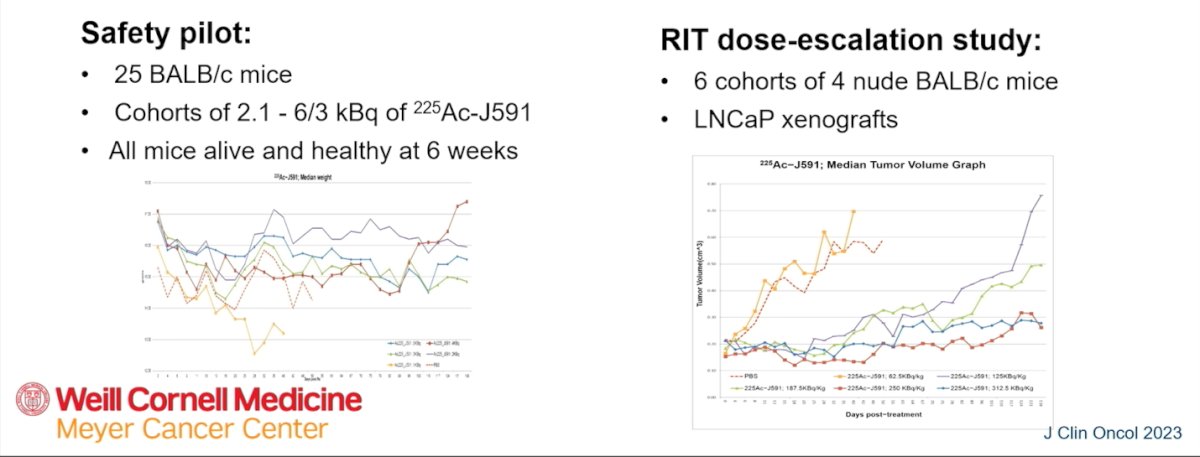
As illustrated below, in a safety pilot, 25 BALB/c mice received escalating doses of Actinium-225–J591, and all remained alive and healthy at six weeks. A subsequent radiopharmaceutical therapy dose-escalation study included six cohorts of nude BALB/c mice bearing LNCaP xenografts. These experiments demonstrated dose-dependent antitumor activity, providing the preclinical foundation for clinical translation.
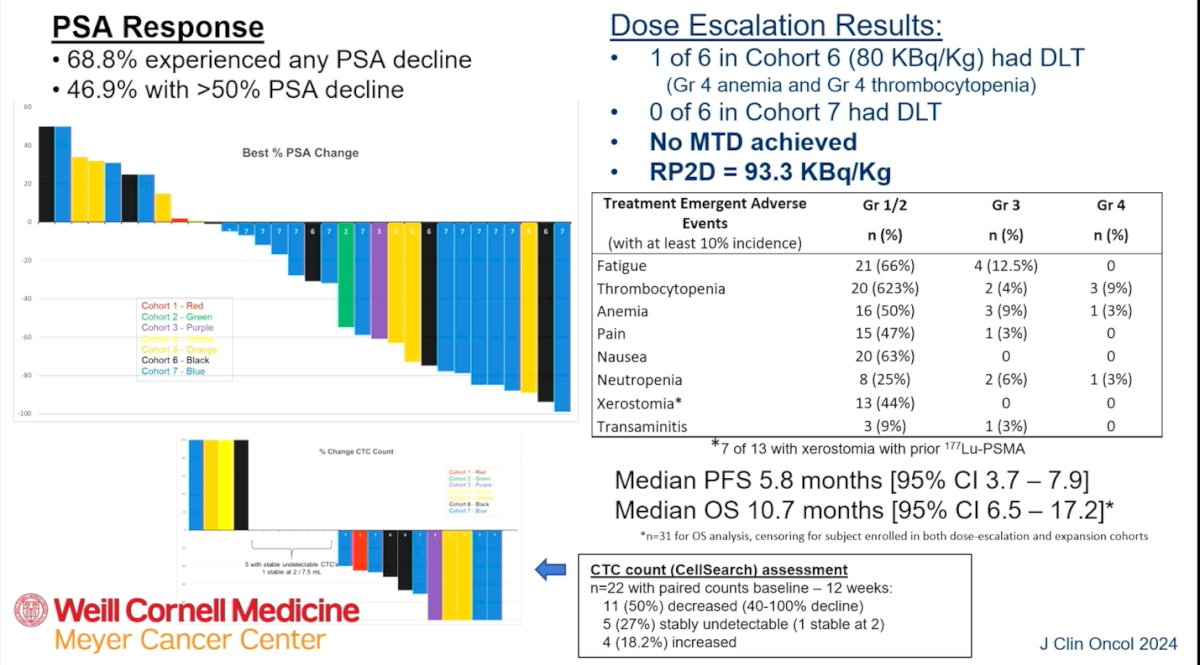
Notably, in the RIT dose-escalation study, 68.8% of patients experienced any PSA decline, with 46.9% achieving a greater than 50% reduction. In the dose-escalation phase, one dose-limiting toxicity was observed in Cohort 6 (80 KBq/kg), consisting of grade 4 anemia and thrombocytopenia, while no DLTs were seen in Cohort 7. No maximum tolerated dose was reached, and the recommended phase II dose was established at 93.3 KBq/kg.
Treatment-emergent adverse events were primarily hematologic, including thrombocytopenia and anemia, along with fatigue and nausea. Grade 4 events were limited. Median progression-free survival was 5.8 months, and median overall survival was 10.7 months. Circulating tumor cell assessments showed that half of the evaluable patients had a decline at 12 weeks, with additional patients maintaining stable undetectable counts, supporting biological activity across multiple response measures.
Dr. Tagawa next reviewed the early clinical development of Actinium-225 J591, highlighting that a single dose of the PSMA-targeted alpha emitter using the intact J591 monoclonal antibody was feasible and associated with generally low-grade, transient toxicity along with signals of preliminary efficacy. The initial dual-center trial was supported through a combination of philanthropic funding and grant support, underscoring the academic-driven nature of the program.1
Building on these results, multiple investigator-initiated studies were launched, including fractionated and multi-cycle dosing strategies, combination approaches with Lutetium-177 PSMA small molecules, and integration with pembrolizumab and androgen receptor inhibitors, as well as a re-treatment pilot. As the program matured, registration-directed development transitioned to a biotechnology partner to support broader clinical advancement.
Dr. Tagawa addressed the additional logistical considerations required for investigator-initiated radiopharmaceutical therapy trials. For the combination of Actinium-225 J591 with Lutetium-177 PSMA small-molecule ligands, the rationale was supported by clinical biology, radiation physics principles, and encouraging preclinical combination data. While there was a pre-existing IND for Actinium-225 J591, separate regulatory planning was required for the Lutetium-177 PSMA component. Rather than pursuing a new IND independently, the team secured sponsorship for drug supply from the POINT trial, and in a subsequent phase, utilized a commercial Lutetium-177 PSMA-617 supply.
For the pembrolizumab plus androgen receptor inhibitor combination, with or without Actinium-225 J591, drug procurement and distribution added another layer of complexity. Pembrolizumab was supplied and shipped to participating sites by the industry, while early participants were treated at Weill Cornell Medicine before expanding to the transportation of radiolabeled investigational product to additional centers. These examples underscore that beyond scientific rationale, manufacturing, regulatory navigation, and coordinated drug distribution are central to successfully executing investigator-initiated RPT trials.
Lastly, Dr. Tagawa reviewed the evolution of PSMA-based strategies within the NCTN framework, beginning in the post-CHAARTED era when ADT plus docetaxel became standard of care. Early concepts presented in 2015 through Alliance and NRG explored combinations such as ADT/docetaxel with or without radium-223, as well as ADT with abiraterone-based intensification strategies. This led to a series of cooperative group studies, including A031601, A031703, and A031804, although some approved protocols were later reshaped by industry partnerships and shifting development priorities. The A031905 study evaluated ADT plus ARPI with or without Lutetium-177 PSMA-617, but growing pharmaceutical interest in global registration trials redirected its trajectory. Ultimately, this work evolved into AFT-53, or PSMAaddition, in addition to evaluating ADT plus ARPI with or without Lutetium-177 PSMA-617, with radiographic progression-free survival as the primary endpoint, mandatory PSMA PET screening, incorporation of crossover, and expansion to six treatment cycles reflecting the maturation of PSMA-directed therapy within national cooperative group research.
Presented by: Scott Tagawa, MD, MS, FACP, FASCO, Professor of Medicine and Urology, Weill Cornell Medicine, New York Presbyterian Hospital, New York City, NY
Written by: Julian Chavarriaga, MD – Urologic Oncologist, Department of Urology at Penn State Health. @chavarriagaj on Twitter during the 2026 American Society for Radiation Oncology (ASTRO) Multidisciplinary Radiopharmaceutical Therapy Symposium (MRPTS) held in Palm Desert, California, between February 17th and 18th, 2026.
Reference: