(UroToday.com) The eighth session of the 2022 International Kidney Cancer Symposium (IKCS): North America meeting focused on regulatory considerations for new trials in advanced renal cell carcinoma (RCC). In this context, Dr. Chang presented on “FDA 101: Illuminating the Black Box”.
Dr. Chang began with some context in terms of the role of the FDA, with responsibility for protecting the public health by ensuring the safety, efficacy, and security of human and veterinary drug, biological products, and medical devices. Further, the FDA is charged with ensuring the safety of the food supply, cosmetics, and products that emit radiation. As a result, the FDA ensures that new drugs and certain biologics have been proven safe and effective prior to marketing. She emphasized that the FDA does not take price into account in its assessment, nor does it regulate “the practice of medicine”.
Historically, the FDA used to require two or more trials for approval. However, now one is acceptable when a second trial is not reasonable such as for life-threatening conditions. Thus, based on legislative mandate, the FDA requires demonstration of efficacy with acceptable safety in adequate and well-controlled studies. Importantly, approvals are not made broadly for a drug, but for a specific indication which defines and appropriate patient population and provides adequate information to inform safe and effective use.
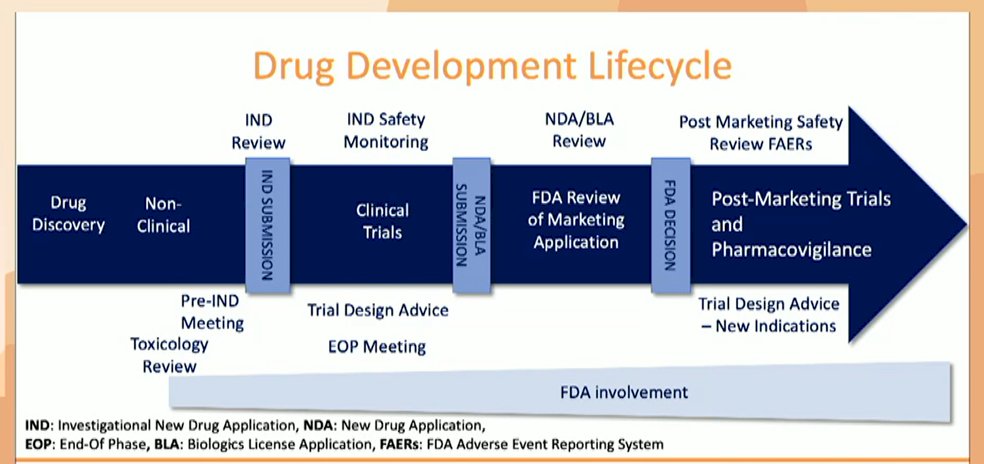
This process involves the entirety of the drug development lifestyle, of which the FDA is involved in the entirety though with increasing involvement in the later stages. An IND is required for all human trials, whether phase I to III. Following completion of a phase III trial, the FDA reviews raw data, which is unique among regulatory agencies. While the figure below highlights many interactions that may be had with the FDA, this list of meetings is not comprehensive, with many other potential reasons at the discretion of either the applicant or the FDA.
Moving forward, Dr. Chang discussed how the FDA examines trial endpoints. She noted that the strength of efficacy endpoints may be assessed across a variety of criteria:
- endpoint selection: ie. what is being measured? In this context, the FDA prioritizes endpoints that show a direct benefit to patients in terms of how they feel, function, or survive.
- measurement characteristics: ie. how accurately the endpoint may be measured. In this context, we need to consider the accuracy with which an endpoint can be measured and the potential susceptibility to bias.
- the magnitude of effect: ie. how much effect on the endpoint is observed? In this context, a larger magnitude of effect may mitigate other weakness in endpoint assessment.
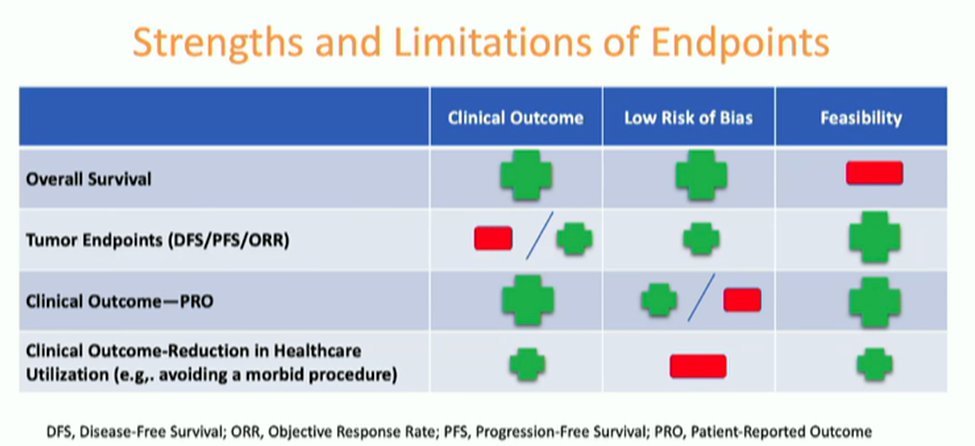
Dr. Chang emphasized that each endpoint has strengths and limitations. Overall survival has long been considered the gold standard endpoint. Certainly, OS minimizes bias due to its ability to be objectively determined and reflects both efficacy and safety. However, trials powered on OS may take a long time to accrue and require larger sample sizes. Additionally, future treatments or cross-over may confound OS outcomes.

Progression-free survival (PFS) is, as Dr. Chang pointed out, a clinically relevant endpoint. It accounts for stable tumor and is not affected but future treatments or cross-over. Additionally, the use of PFS allows for smaller trials and shorter durations of follow-up. However, there are circumstances where outcome adjudication may be difficult and single arm PFS is often not interpretable.

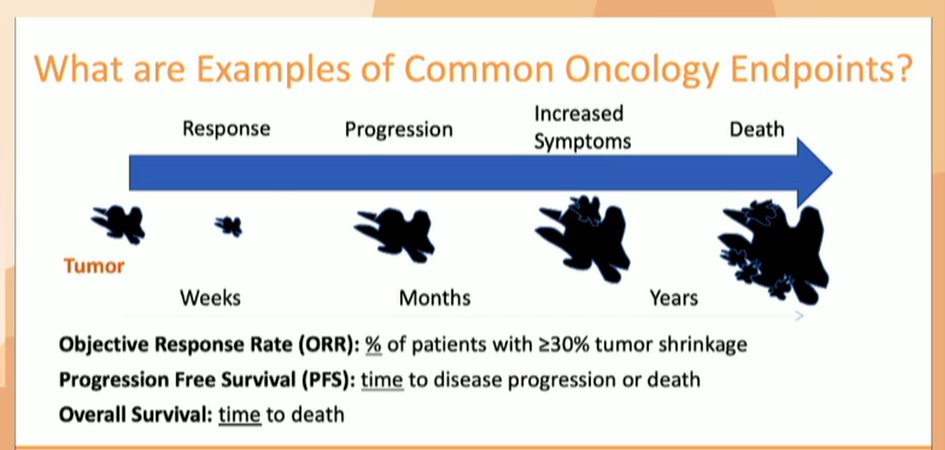
Finally, Dr. Chang discussed objective response rate (ORR), which she noted is particularly relevant for rare disease where large trials are hard to accrue. However, there may be considerable uncertainty regarding the clinical benefit of agents when ORR is utilized.

Dr. Chang then highlighted the overall balance of strengths and limitations of each of these endpoints, along with patient reported clinical outcomes and reduction of health care resource utilization, accounting for clinical outcomes, risk of bias, and feasibility. Clearly, there is a balance between each of these characteristics.
While patient-reported outcome metrics are critical in understanding the benefits and toxicity of any given agent, she noted that there may be issues with sensitivity of PRO measures. Particularly, for patients with early-stage disease, many patients are asymptomatic. Thus, time to symptomatic deterioration is typically utilized. Dr. Chang noted that increasingly trials supporting regulatory approval are open-label with PROs collected as secondary outcomes in many relevant trials. She noted that PRO data are critical for a complete assessment of safety and tolerability and emphasized that this should be done in nearly all trials. Whether used for safety or efficacy assessment, she noted that PRO endpoint should have clearly stated and well-defined objectives.
She noted that, when considering new endpoints, we need to balance the certainty of data regulatory burden that we impose.
In conclusion, Dr. Chang noted that the FDA is involved in the entire drug development lifecycle and that each endpoint that may be used has strengths and limitations. In the context of each trial, the appropriateness of an endpoint depends on multiple factors including the patient population, measurement characteristics, and type of trial.