In this important classification every entity was described taking into account not only the pathological features, but also the genetic characteristics. Moreover, some tumors have been defined as benign (tubolo-papillary adenoma, oncocytoma and metanephric adenoma) and sarcomatoid carcinoma has been recognized as a separate entity rather than an extreme degree of dedifferentiation common to all other histologic types.
In the last decade, new neoplasms have been identified and described; therefore the International Society of Urological Pathology (ISUP) decided to organize a new consensus conference that was held in March 2012 in Vancouver, Canada. The goal was to delineate the recommendations regarding new renal cell neoplasms and emerging entities, as well as new concepts and clarifications of the renal tumors already described in the current WHO classification. The methodology consisted of a thorough revision of the literature, a survey, and a plenary discussion during which the participants were asked to vote for each specific question.
Several new renal cell tumors were proposed by the ISUP in Vancouver. These included tubulocystic renal cell carcinoma (RCC), RCC associated with acquired cystic kidney disease, clear cell papillary RCC, t(6;11) translocation RCC with consequent reclassification of the entire group of tumors with translocation as “MiT Family Translocation Renal Cell Carcinoma”, hereditary leiomyomatosis and RCC (HLRCC)-associated RCC and, finally, succinate dehydrogenase-deficient renal carcinoma.
Tubulocystic RCC (Figure 1) appears on imaging as a complex cyst type 3-4 according to Bosniak criteria. It usually appears in males with a mean age of 60 years. Grossly, it is well circumscribed with a translucent cut surface and composed by multiple cysts. Microscopically, it is composed by tubules of variable size lined by a single layer of cells with eosinophilic cytoplasm and round nuclei with nucleoli. These cells are usually positive for AMACR immunohistochemical staining and present trisomy of chromosomes 7 and 17, similar to papillary RCC.
RCC associated with acquired cystic kidney disease (Figure 2) represents the histologic type most frequently occurring in end stage renal disease. It is composed by voluminous eosinophilic cells that may show a cribriform pattern with oxalate crystals. These neoplastic cells stain positively for AMACR and are characterized by numeric chromosomal alterations.
Clear cell papillary RCC is a neoplasm originally described in the setting of end stage renal disease, and also recognized in otherwise normal renal parenchyma and represents 4% of all renal tumors. It is composed by clear cells forming papillae and tubules that are characterized by a typical nuclear arrangement away from the basal membrane. These tumors are negative for AMACR; moreover, they lack any genetic alteration of chromosomes 3, 7 and 17.
RCC with t(6;11) translocation, similarly to other renal carcinomas with translocation, had been initially described in younger individuals but has also been demonstrated in older patients. They show a peculiar morphology with psudoresette-like structures and are consistently positive for HMB45 and cathepsin K.
Succinate dehydrogenase-deficient renal carcinoma (Figure 3) is a rare entity reported in patients with germline mutation of succinate-dehydrogenase gene. Mutation in one of SDH subunit (A, B, C and D) predispose not only to RCC but also to paraganglioma/pheocromocytoma, gastrointestinal stromal tumor and pituitary adenoma. Many of SDH-deficient RCCs have been microscopically characterized by sheets of uniform eosinophilic cells with flocculent cytoplasmic inclusions. To identify these tumors, loss of immunohistochemical staining for succinate dehydrogenase subunit B (SDHB) is considered a reliable tool.
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC)-associated RCC is a neoplasm made of cells with abundant eosinophilic cytoplasm with nuclei surrounded by a perinuclear halo with prominent nucleoli. This neoplasm occurs in either the aforementioned syndrome or in the sporadic setting. It is an aggressive tumor, characterized by germline mutations in the gene coding for the enzyme fumarate hydratase, located on chromosome 1q42, and the loss of immunohistochemical expression can be used as diagnostic tool.
Figure 1:

Figure 2:
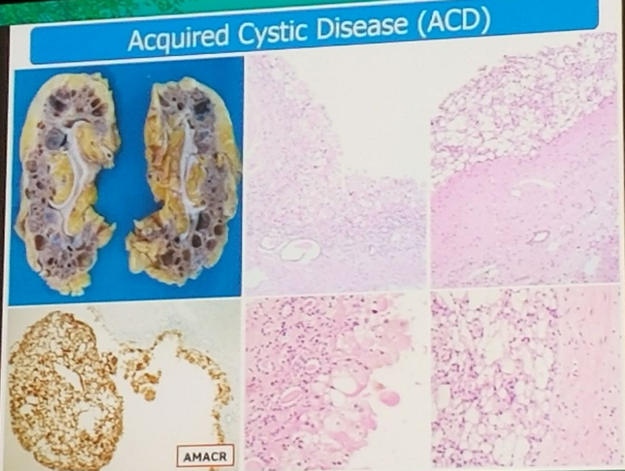
Figure 3:

Speaker: Guido Martignoni, Department of Pathology and Diagnostic, University of Verona, Verona, Italy
Written By: Hanan Goldberg, MD, Urologic Oncology Fellow (SUO), University of Toronto, Princess Margaret Cancer Centre @GoldbergHanan at The 15th Meeting of the EAU Section of Oncological Urology ESOU18 - January 26-28, 2018 - Amsterdam, The Netherlands