(UroToday.com) On the third day of the American Society for Clinical Oncology (ASCO) Genitourinary Cancer Symposium 2022 in a symposium focused on biomarkers and therapies in renal cell carcinoma (RCC), Dr. Dean presented on germline mutations of interest in renal cell carcinoma (RCC).
Dr. Dean began by highlighting hereditary disease syndromes that include renal cell carcinoma. The first, and best recognized of these, is von Hippel-Lindau (VHL) syndrome. This autosomal dominant condition, involving the VHL gene (located at 3p25.3), has a mean age of diagnosis of 26 years and affects on in every 36,000 live births. Further, VHL may be divided into type 1 which does not include pheochromocytoma (representing approximately 80% of cases) and type 2 which does (representing approximately 20% of cases). These subtypes of VHL have dramatically different underlying biology: Dr. Dean emphasized that type I VHL is predominantly associated with loss-of-function mutations while type 2 VHL is nearly entirely associated with missense mutations.

He further highlighted the timeline of a typical VHL patient, moving from an initial diagnosis at age 26 to the first diagnosis of renal cell carcinoma (RCC) at age 44 and increasing risks of RCC over time reaching 70% by age 60.
Work into the underlying mechanisms of clear cell renal cell carcinogenesis in VHL lead to critical insights in the mechanisms of all clear cell renal cell carcinoma. VHL binds to HIF-1alpha, regulating the hypoxia response. Loss of VHL function leads to uncontrolled expression of genes, including those related to angiogenesis, glucose transport and cellular growth, that are normally only expressed during hypoxia.

Dr. Dean then highlighted work from Dr. Knudson who described the two-hit hypothesis in which a germline/inherited mutation is present on one allele and a second somatic mutation or deletion is required on the other allele to lead to tumor growth. In sporadic tumors, the same tumor suppressor gene is often mutated. In most sporadic clear cell renal cell carcinoma, the VHL gene is mutated or methylated.
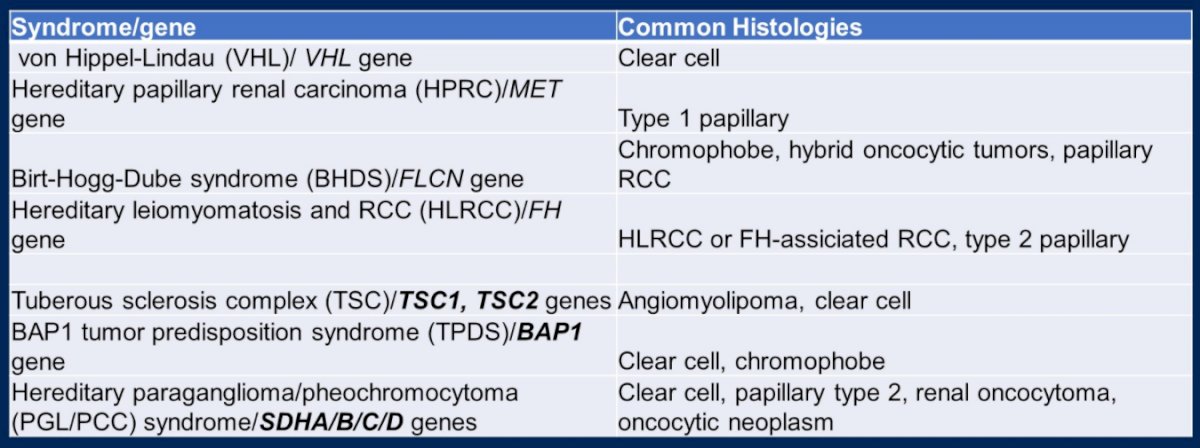
Dr. Dean then discussed other hereditary cancer syndromes involving renal cell carcinoma including hereditary papillary RCC, hereditary leiomyomatosis and renal cell carcinoma, and Birt-Hogg-Dube which is associated with chromophobe RCC.
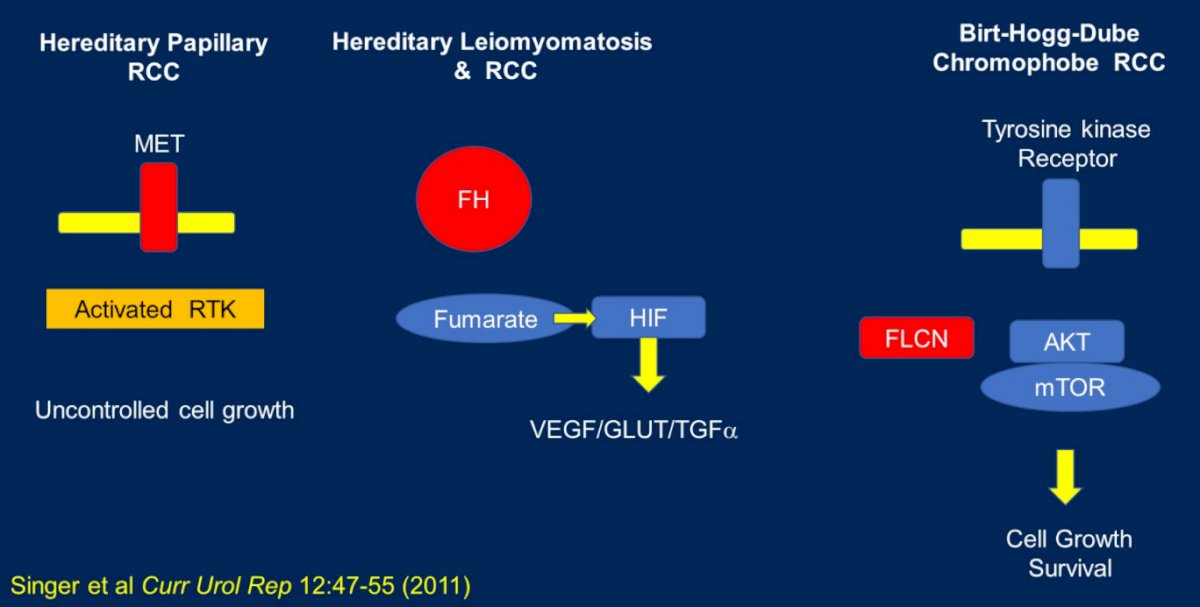
In addition to these syndromes which have been recognized for quite some time, the National Comprehensive Cancer Network (NCCN) now recognizes seven additional hereditary kidney cancer genes including TSC1, TSC2, BAP1, SDHA, SDHB, and SDHC.
Dr. Dean then highlighted criteria, based on the NCCN guidance, that may be used as criteria for further genetic risk evaluation including:
- a close relative with a known pathogenic variant in a cancer susceptibility gene
- diagnosis <47 years of age
- bilateral or multifocal tumors
- one or more first or second degree relatives with RCC
- multifocal papillary histology
- HLRCC-associated RCC
- BHDS-related histology
- angiomyolipomas of the kidney with one or more TSCC feature
- SDH-deficient RCC histology.
Based on significant from the Renal Cell Carcinoma Genome-Wide Association Studies (GWASs), sixteen independent risk loci have been identified including some what are sex-specific and many of which involve hypoxia related genes and the PI3K/AKT pathway.
In conclusion, Dr. Dean emphasized that there are many genes causing inherited kidney cancer, though syndromes are relatively rare. Identifying patients and families can improve cancer outcomes through early diagnosis and improved disease management.
Presented by: Michael Dean, PhD, National Cancer Institute